Муковисцидоз
Last reviewed: 26.06.2018

Avem reguli stricte de aprovizionare și oferim linkuri doar către site-uri medicale reputate, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale evaluate de colegi. Rețineți că numerele din paranteze ([1], [2] etc.) sunt linkuri către aceste studii pe care se poate da clic.
Dacă considerați că vreunul dintre conținuturile noastre este inexact, învechit sau altfel discutabil, vă rugăm să îl selectați și să apăsați Ctrl + Enter.
Fibroza chistică este o boală genetică autosomal recesivă, monogenică, caracterizată printr-o tulburare a secreției glandelor exocrine ale organelor vitale, cu afectare în principal a sistemelor respirator și digestiv, cu evoluție severă și prognostic nefavorabil.
 [ 1 ]
[ 1 ]
Epidemiologie
Incidența fibrozei chistice fluctuează între 1:2.500 și 1:4.600 de nou-născuți. În fiecare an, se nasc aproximativ 45.000 de persoane cu fibroză chistică în întreaga lume. Incidența purtătorilor genei fibrozei chistice este de 3-4%, aproximativ 275 de milioane de oameni din întreaga lume fiind purtători ai acestei gene, dintre care aproximativ 5 milioane locuiesc în Rusia și aproximativ 12,5 milioane în țările CSI.
Cauze fibroză chistică
Fibroza chistică se transmite pe cale autosomal recesivă. Gena fibrozei chistice este situată în autosomul 7, conține 27 de exoni și este alcătuită din 250.000 de perechi de nucleotide.
O singură genă poate avea mai multe mutații, fiecare dintre acestea fiind specifică unei anumite populații sau regiuni geografice. Au fost descrise peste 520 de mutații, cea mai frecventă fiind delta-P-508, adică o substituție a aminoacidului fenilalanină la poziția 508.
Patogeneza
Mutațiile genei fibrozei chistice perturbă structura și funcția unei proteine numite CFTR (regulator transmembranar al fibrozei chistice). Această proteină acționează ca un canal de clorură implicat în schimbul apă-electroliți al celulelor epiteliale ale sistemului bronhopulmonar, tractului gastrointestinal, pancreasului, ficatului și sistemului reproducător. Ca urmare a perturbării funcției și structurii proteinei CFTR, ionii de clorură Cl⁻ se acumulează în interiorul celulei. Aceasta duce la o modificare a potențialului electric din lumenul canalelor excretoare, ceea ce facilitează fluxul unor cantități mari de ioni de sodiu (Na⁺ ) din lumenul canalului în celulă și sporește în continuare absorbția apei din spațiul pericelular.
Ca urmare a acestor modificări, secreția majorității glandelor exocrine se îngroașă, evacuarea acesteia este perturbată, ceea ce duce la tulburări secundare pronunțate în organe și sisteme, cele mai pronunțate în sistemele bronhopulmonar și digestiv.
În bronhii se dezvoltă un proces inflamator cronic de intensitate variabilă, funcția epiteliului ciliat este brusc perturbată, sputa devine foarte vâscoasă, groasă, foarte dificil de evacuat, se observă stagnarea acesteia, se formează bronhiolo- și bronșiectazii, care în timp devin mai frecvente. Aceste modificări duc la o creștere a hipoxiei și la formarea cardiopatiei pulmonare cronice.
Pacienții cu fibroză chistică sunt extrem de predispuși la dezvoltarea inflamației cronice în sistemul bronhopulmonar. Acest lucru se datorează perturbărilor pronunțate ale sistemului local de apărare bronhopulmonară (niveluri reduse de IgA, interferon, funcție fagocitară a macrofagelor alveolare și a leucocitelor).
Macrofagele alveolare joacă un rol major în dezvoltarea inflamației cronice în sistemul bronhopulmonar. Acestea produc cantități mari de IL-8, care crește dramatic chemotaxia neutrofilelor în arborele bronșic. Neutrofilele se acumulează în cantități mari în bronhii și, împreună cu celulele epiteliale, secretă numeroase citokine proinflamatorii, inclusiv IL-1, 8, 6, factorul de necroză tumorală și leucotriene.
Un rol important în patogeneza afectării sistemului bronhopulmonar îl joacă și activitatea ridicată a enzimei elastază. Se face distincție între elastaza exogenă și endogenă. Prima este produsă de flora bacteriană (în special Pseudomonas aeruginosa), a doua - de leucocitele neutrofile. Elastaza distruge epiteliul și alte elemente structurale ale bronhiilor, ceea ce contribuie la perturbarea suplimentară a transportului mucociliar și la formarea rapidă a bronșiectaziilor.
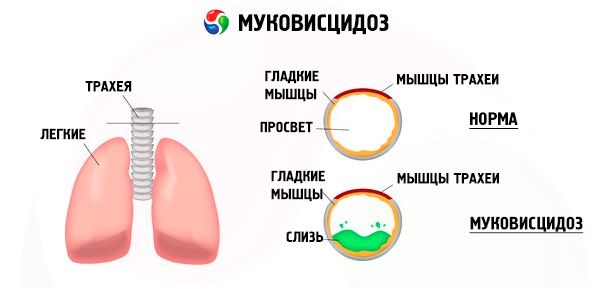
Leucocitele neutrofile secretă și alte enzime proteolitice. Alfa-1-antipirina și inhibitorul secretor al leucoproteazelor contracarează influența enzimelor proteolitice și, prin urmare, protejează sistemul bronhopulmonar de influența lor dăunătoare. Cu toate acestea, din păcate, la pacienții cu fibroză chistică, acești factori de protecție sunt suprimați de o cantitate semnificativă de protează neutrofilă.
Toate aceste circumstanțe contribuie la introducerea infecției în sistemul bronhopulmonar și la dezvoltarea bronșitei purulente cronice. În plus, trebuie luat în considerare faptul că proteina defectă codificată de gena fibrozei chistice modifică starea funcțională a epiteliului bronșic, ceea ce favorizează aderența bacteriilor la epiteliul bronșic, în principal Pseudomonas aeruginosa.
Pe lângă patologia sistemului bronhopulmonar, fibroza chistică provoacă și leziuni severe pancreasului, stomacului, intestinului gros și subțire și ficatului.
Simptome fibroză chistică
Fibroza chistică se manifestă prin diverse simptome clinice. La nou-născuți, boala se poate manifesta prin ileus meconial. Din cauza lipsei sau chiar a absenței complete a tripsinei, meconiul devine foarte dens, vâscos și se acumulează în regiunea ileocecală. În plus, se dezvoltă obstrucția intestinală, care se manifestă prin vărsături intense cu amestec de bilă, distensie abdominală, lipsă de excreție de meconiu, dezvoltarea simptomelor de peritonită și creșterea rapidă a manifestărilor clinice ale sindromului de intoxicație severă. Copilul poate deceda în primele zile de viață dacă nu se efectuează o intervenție chirurgicală urgentă.
În cazurile mai puțin severe, un semn caracteristic al fibrozei chistice este scaunul abundent, frecvent, onctuos, cu o cantitate mare de grăsime, cu un miros foarte neplăcut. La 1/3 dintre pacienți se observă prolapsul rectului.
Ulterior, pacienții continuă să prezinte disfuncții intestinale, sindrom de malabsorbție, tulburări severe de dezvoltare fizică și hipovitaminoză severă.
În primul sau al doilea an de viață apar simptome de afectare a sistemului bronhopulmonar (formă ușoară a bolii), care se manifestă printr-o tuse care poate fi extrem de pronunțată și seamănă cu o tuse cu tuse convulsivă. Tusea este însoțită de cianoză, dificultăți de respirație și separarea sputei groase, inițial mucoasă, apoi purulentă. Treptat, se formează un tablou clinic de bronșită obstructivă cronică și bronșiectazie, emfizem pulmonar și insuficiență respiratorie. Copiii sunt extrem de susceptibili la infecții virale și bacteriene respiratorii acute, ceea ce contribuie la exacerbări și progresia patologiei bronhopulmonare. Este posibilă dezvoltarea astmului bronșic dependent de infecție.
La copiii de vârstă școlară, fibroza chistică se poate manifesta ca „colică intestinală”. Pacienții se plâng de dureri abdominale paroxistice severe, balonare și vărsături repetate. La palparea abdomenului, se determină formațiuni dense, situate în proiecția intestinului gros - mase fecale amestecate cu mucus gros și dens. Copiii sunt foarte predispuși la dezvoltarea alcalozei hipocloremice din cauza excreției excesive de sare odată cu transpirația pe vreme caldă, în timp ce pe pielea copilului apare „îngheț sărat”.
Tulburări ale sistemului bronhopulmonar la adulți
Afectarea sistemului bronhopulmonar la pacienții cu fibroză chistică (forma pulmonară a bolii) se caracterizează prin dezvoltarea bronșitei obstructive purulente cronice, bronșiectaziei, pneumoniei cronice, emfizemului pulmonar, insuficienței respiratorii și cardiopatiei pulmonare. Unii pacienți dezvoltă pneumotorax și alte complicații ale fibrozei chistice: atelectazie, abcese pulmonare, hemoptizie, hemoragie pulmonară și astm bronșic dependent de infecție.
Pacienții se plâng de o tuse paroxistică dureroasă, cu spută mucopurulentă foarte vâscoasă, dificil de separat, uneori cu un amestec de sânge. În plus, dificultățile de respirație sunt extrem de caracteristice, mai întâi în timpul efortului fizic, apoi în repaus. Dificultățile de respirație sunt cauzate de obstrucția bronșică. Mulți pacienți se plâng de rinită cronică cauzată de polipoză și sinuzită. Sunt caracteristice și slăbiciune semnificativă, scăderea progresivă a performanței, boli virale respiratorii acute frecvente. La examinare, se atrage atenția asupra pielii palide, umflăturii feței, cianozei membranelor mucoase vizibile și dificultăți severe de respirație. Odată cu dezvoltarea cardiopatiei pulmonare decompensate, apare edemul la nivelul picioarelor. Se poate observa îngroșarea falangelor terminale ale degetelor sub formă de copane și a unghiilor sub formă de sticle de ceas. Toracele capătă o formă de butoi (datorită dezvoltării emfizemului pulmonar).
Percuția plămânilor relevă semne de emfizem pulmonar - un sunet de cutie, o limitare bruscă a mobilității marginii pulmonare și o coborâre a marginii inferioare a plămânilor. Auscultația plămânilor relevă respirație dificilă cu expirație prelungită, respirație șuierătoare uscată dispersată și respirație șuierătoare umedă, medie și fină, cu bule. În cazul emfizemului pulmonar sever, respirația este puternic slăbită.
Manifestări extrapulmonare ale fibrozei chistice
Manifestările extrapulmonare ale fibrozei chistice pot fi destul de pronunțate și apar frecvent.
Leziuni pancreatice
Insuficiența funcției exocrine a pancreasului, de severitate variabilă, se observă la 85% dintre pacienții cu fibroză chistică. În cazul leziunilor minore ale pancreasului, sindroamele de maldigestie și malabsorbție sunt absente, existând doar manifestări de laborator ale insuficienței exocrine (niveluri scăzute de tripsină și lipază în sânge și conținutul duodenal; adesea steatoree severă). Se știe că pentru a preveni sindromul de maldigestie, este suficientă o secreție de doar 1 până la 2% din lipaza totală. Clinic se manifestă doar tulburări semnificative ale funcției exocrine.
În condiții normale, acinii pancreatici produc o secreție lichidă bogată în enzime. Pe măsură ce secreția se deplasează de-a lungul canalului excretor, aceasta se îmbogățește cu apă și anioni și devine și mai lichidă. În fibroza chistică, din cauza unei tulburări în structura și funcția regulatorului transmembranar (canalul de clorură), secreția pancreatică nu primește o cantitate suficientă de lichid, devine vâscoasă, iar viteza deplasării sale de-a lungul canalului excretor încetinește brusc. Proteinele secreției se depun pe pereții canalelor excretoare mici, rezultând obstrucția acestora. Pe măsură ce boala progresează, se dezvoltă în cele din urmă distrugerea și atrofia acinilor - se formează pancreatită cronică cu insuficiență pancreatică exocrină. Acest lucru se reflectă clinic în dezvoltarea sindroamelor de maldigestie și malabsorbție. Insuficiența pancreatică este principala cauză a malabsorbției de grăsimi în fibroza chistică, dar aceasta se observă de obicei cu un deficit semnificativ de lipază. Forsher și Durie (1991) indică faptul că în absența completă a lipazei pancreatice, grăsimea este descompusă și absorbită în proporție de 50-60%, ceea ce se datorează prezenței lipazelor gastrice și salivare (sublinguale), a căror activitate este aproape de limita inferioară a normei. Odată cu perturbarea descompunerii și absorbției grăsimilor, există o perturbare a descompunerii și reabsorbției proteinelor. Aproximativ 50% din proteinele primite prin alimente se pierd prin fecale. Absorbția carbohidraților este afectată într-o măsură mai mică, în ciuda deficitului de α-amilază, dar metabolismul carbohidraților poate fi perturbat semnificativ.
Lezarea pancreasului se manifestă prin dezvoltarea sindromului de maldigestie și malabsorbție, cu pierdere semnificativă în greutate și scaune abundente cu grăsime.
Dezvoltarea sindroamelor de maldigestie și malabsorbție este, de asemenea, facilitată de disfuncția severă a glandelor intestinale, secreția afectată de suc intestinal și scăderea conținutului de enzime intestinale din acesta.
Sindroamele de maldigestie și malabsorbție sunt numite și forma intestinală a fibrozei chistice.
Funcția endocrină afectată a pancreasului (diabetul zaharat) este observată la pacienții cu fibroză chistică în stadiile avansate ale bolii (la 2% dintre copii și 15% dintre adulți)
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Afectarea ficatului și a tractului biliar
La 13% dintre pacienții cu forme mixte și intestinale de fibroză chistică se dezvoltă ciroză hepatică. Aceasta este cea mai tipică pentru mutațiile W128X, delta-P508 și X1303K. Ciroza biliară hepatică cu hipertensiune portală este detectată la 5-10% dintre pacienți. Conform lui Welch, Smith (1995), semnele clinice, morfologice, de laborator și instrumentale ale afectării hepatice sunt detectate la 86% dintre pacienții cu fibroză chistică.
Mulți pacienți cu fibroză chistică dezvoltă și colecistită cronică, adesea calculoasă.
Disfuncția glandelor sexuale
Pacienții cu fibroză chistică pot prezenta azoospermie, care este cauza infertilității. Fertilitatea redusă este, de asemenea, tipică la femei.
Etape
Există trei grade de severitate a fibrozei chistice pulmonare.
Forma ușoară de fibroză chistică se caracterizează prin exacerbări rare (nu mai mult de o dată pe an); în perioadele de remisie, manifestările clinice sunt practic absente, iar pacienții sunt capabili să lucreze.
Severitate moderată - exacerbările se observă de 2-3 ori pe an și durează aproximativ 2 luni sau mai mult. În faza de exacerbare, există o tuse intensă cu spută dificil de separat, dificultăți de respirație chiar și cu efort fizic minor, temperatură corporală subfebrilă, slăbiciune generală, transpirații. În același timp, există o încălcare a funcției exocrine a pancreasului. În faza de remisie, capacitatea de lucru nu este complet restabilită, dificultățile de respirație în timpul efortului fizic persistă.
Cursul sever este caracterizat prin exacerbări foarte frecvente ale bolii. Remisiile sunt practic absente. În tabloul clinic, se evidențiază insuficiență respiratorie severă, simptome de cardiopatie pulmonară cronică, adesea decompensată, hemoptizia este tipică. Se observă o pierdere semnificativă în greutate, pacienții sunt complet invalidi. De regulă, patologia bronhopulmonară severă este însoțită de o disfuncție accentuată a pancreasului.
Formulare
- Leziuni bronhopulmonare
- Pneumonie repetată și recurentă cu evoluție prelungită.
- Pneumonie abcesantă, în special la sugari.
- Pneumonie cronică, în special bilaterală.
- Astmul bronșic refractar la terapia tradițională.
- Bronșită recurentă, bronșiolită, în special cu cultură de Pseudomonas aeruginosa.
- Modificări ale tractului gastrointestinal
- Ileus meconial și echivalenții săi.
- Sindromul de absorbție intestinală afectată de origine necunoscută.
- Icter obstructiv la nou-născuți cu evoluție prelungită.
- Ciroză hepatică.
- Diabetul zaharat.
- Refluxul gastroesofagian.
- Colelitiază.
- Prolaps rectal.
- Modificări ale altor organe și sisteme
- Tulburări de creștere și dezvoltare.
- Dezvoltare sexuală întârziată.
- Infertilitate masculină.
- Polipi nazali.
- Frați din familii cu fibroză chistică.
[ 24 ]
Complicații și consecințe
Complicațiile din tractul gastrointestinal includ:
- Diabetul zaharat se dezvoltă la 8-12% dintre pacienții cu vârsta peste 25 de ani.
- Colonopatie fibrozantă.
- Ileus meconial în perioada neonatală (la 12% dintre nou-născuții cu fibroză chistică), sindrom de obstrucție intestinală distală, prolaps rectal, ulcer peptic și reflux gastroesofagian.
Complicații hepatice:
- Steatoză hepatică (la 30-60% dintre pacienți),
- Ciroză biliară focală, ciroză biliară multinodulară și hipertensiune portală asociată.
Hipertensiunea portală duce uneori la deces din cauza varicelor esofagiene.
Prevalența colecistitei și a calculilor biliari este mai mare la pacienții cu fibroză chistică decât la alte persoane.
Pubertate întârziată și fertilitate scăzută și alte complicații. Majoritatea bărbaților au azoospermie și subdezvoltare a canalului deferent.
Diagnostice fibroză chistică
Analiză generală de sânge - anemia de severitate variabilă este tipică, de obicei normo- sau hipocromă. Anemia are o geneză polifactorială (absorbție redusă a fierului și vitaminei B12 în intestin din cauza dezvoltării sindromului de malabsorbție). Leucopenia este posibilă, cu dezvoltarea bronșitei purulente și a pneumoniei - leucocitoză, VSH crescută.
Analiza generală a urinei - fără modificări semnificative, în cazuri rare se observă o ușoară proteinurie.
Examen coprologic - se observă steatoree, creatioree. Becker (1987) recomandă măsurarea chimotripsinei și a acizilor grași din fecale. Înainte de a determina chimotripsina în fecale, este necesar să se întrerupă administrarea enzimelor digestive cu cel puțin 3 zile înainte de examinare. În fibroza chistică, cantitatea de chimotripsină din fecale este redusă, iar cantitatea de acizi grași este crescută (excreția normală de acizi grași este mai mică de 20 mmol/zi). Este necesar să se țină cont de faptul că o excreție crescută de acizi grași prin fecale se observă și în:
- deficit de acizi grași conjugați în intestinul subțire din cauza insuficienței hepatice, obstrucției căilor biliare, colonizării bacteriene semnificative a intestinului subțire (în acest caz, are loc hidroliza intensivă a acizilor biliari);
- ileită;
- boala celiacă (cu dezvoltarea sindromului de malabsorbție);
- enterită;
- limfoame intestinale;
- Boala Whipple;
- alergii alimentare;
- tranzitul accelerat al maselor alimentare în diareea de diverse origini, sindromul carcinoid, tireotoxicoza.
Test biochimic de sânge - scăderea nivelului total de proteine și albumină, creșterea nivelului de alfa2 și gama globuline, bilirubină și aminotransferaze (în caz de afectare hepatică), scăderea activității amilazei, lipazei, tripsinei și a nivelului de fier și calciu (în cazul dezvoltării sindromului de maldigestie, malabsorbție).
Analiza sputei - prezența unui număr mare de leucocite neutrofile și microorganisme (în timpul bacterioscopiei sputei).
Un studiu al funcției de absorbție a intestinului subțire și al funcției exocrine a pancreasului relevă perturbări semnificative.
Examinarea cu raze X a plămânilor - relevă modificări a căror severitate depinde de severitatea și faza bolii. Cele mai caracteristice modificări sunt:
- creșterea patternului pulmonar din cauza modificărilor interstițiale peribronșice;
- expansiunea rădăcinilor plămânilor;
- imagine a atelectaziei lobulare, subsegmentare sau chiar segmentare a plămânilor;
- creșterea transparenței câmpurilor pulmonare, în principal în secțiunile superioare, poziția joasă și mobilitatea insuficientă a diafragmei, expansiunea spațiului retrosternal (manifestarea emfizemului pulmonar);
- infiltrarea segmentară sau polisegmentară a țesutului pulmonar (în dezvoltarea pneumoniei).
Bronhografia evidențiază modificări cauzate de obstrucția bronhiilor de către sputa vâscoasă (fragmentarea umplerii bronhiilor cu contrast, contururi neuniforme, fenomenul de ruptură bronșică, o scădere semnificativă a numărului de ramuri laterale), precum și bronhoecgazii (cilindrici, mixte), localizați în principal în părțile inferioare ale plămânilor.
Bronhoscopia evidențiază bronșită purulentă difuză cu spută abundentă, groasă și vâscoasă și pelicule fibrinoase.
Spirometria - deja în stadiile incipiente ale bolii relevă insuficiență respiratorie de tip obstructiv (scăderea CVF, FEV1, indicele Tiffno), restrictiv (scăderea CVF) sau, cel mai adesea, obstructiv-restrictiv (scăderea CVF, FEV1, indicele Tiffno).
Testul de transpirație Gibson și Cook (testul electrolitic al transpirației) implică stimularea transpirației folosind electroforeza cu pilocarpină cu determinarea ulterioară a clorurilor din transpirație. Doerehuk (1987) descrie testul astfel. Electroforeza cu pilocarpină se efectuează pe antebraț, curentul electric fiind de 3 mA. După curățarea pielii cu apă distilată, transpirația se colectează folosind hârtie de filtru plasată pe zona stimulată, acoperită cu tifon pentru a preveni evaporarea din aceasta. După 30-60 de minute, hârtia de filtru se îndepărtează și se eluează în apă distilată. Se măsoară cantitatea de transpirație colectată. Pentru a obține rezultate fiabile, este necesar să se colecteze cel puțin 50 mg (de preferință 100 mg) de transpirație.
Dacă concentrația de clorură este mai mare de 60 mmol/l, diagnosticul de fibroză chistică este considerat probabil; dacă concentrația de clorură este mai mare de 100 mmol/l, acesta este fiabil; în acest caz, diferența dintre concentrația de clor și sodiu nu trebuie să depășească 8-10 mmol/l. Hadson (1983) recomandă ca, dacă conținutul de sodiu și clorură din transpirație este la limită, să se efectueze un test cu prednisolon (5 mg oral timp de 2 zile, urmat de determinarea electroliților din transpirație). La persoanele care nu suferă de fibroză chistică, nivelul de sodiu din transpirație scade până la limita inferioară a normalului; în fibroza chistică, acesta nu se modifică. Un test de transpirație este recomandat fiecărui copil cu tuse cronică.
Analiza probelor de sânge sau a probelor de ADN pentru depistarea mutațiilor majore ale genei fibrozei chistice este cel mai sensibil și specific test de diagnostic. Cu toate acestea, această metodă este potrivită doar pentru țările în care rata mutației delta-P508 este mai mare de 80%. În plus, tehnica este foarte scumpă și complexă din punct de vedere tehnic.
Diagnosticul prenatal al fibrozei chistice se efectuează prin determinarea izoenzimelor fosfatazei alcaline din lichidul amniotic. Această metodă devine posibilă de la 18-20 de săptămâni de sarcină.
Principalele criterii pentru diagnosticarea fibrozei chistice sunt următoarele:
- indicații în anamneza dezvoltării fizice întârziate în copilărie, boli respiratorii cronice recurente, tulburări dispeptice și diaree, prezența fibrozei chistice la rude apropiate;
- bronșită obstructivă cronică, adesea recidivantă, cu dezvoltarea de bronșiectazie și emfizem pulmonar, pneumonie adesea recurentă;
- pancreatită cronică recurentă cu o scădere marcată a funcției exocrine, sindrom de malabsorbție;
- conținut crescut de clor în transpirația pacientului;
- infertilitate cu funcție sexuală prezervată.
Diagnosticul cu succes și diagnosticul diferențial al fibrozei chistice sunt facilitate de identificarea grupurilor de risc.
Programul de screening pentru fibroza chistică
- Analize generale de sânge, urină, spută.
- Analiza bacteriologică a sputei.
- Analiza coprologică.
- Test biochimic de sânge: determinarea proteinelor totale și a fracțiilor proteice, glucozei, bilirubinei, aminotransferazelor, fosfatazei alcaline, gama-glutamil transpeptidazelor, potasiului, calciului, fierului, lipazei, amilazei, tripsinei.
- Studiul funcției exocrine a pancreasului și al funcției de absorbție a intestinului.
- Fluoroscopia și radiografia plămânilor, tomografia computerizată a plămânilor.
- ECG.
- Ecocardiografie.
- Bronhoscopie și bronhografie.
- Spirometrie.
- Testul de transpirație.
- Consultație cu un genetician.
- Analiza probelor de sânge sau a probelor de ADN pentru mutații majore ale genei fibrozei chistice.
Cum să examinăm?
Ce teste sunt necesare?
Cine să contactați?
Tratament fibroză chistică
Tipul și severitatea simptomelor fibrozei chistice pot varia foarte mult, așadar nu există un plan de tratament tipic; acesta este individualizat pentru fiecare individ.
Terapia constă în următoarele măsuri terapeutice:
- Exercițiile de respirație și drenajul postural ajută la eliminarea mucusului gros care se acumulează în plămâni. Unele tehnici de curățare a căilor respiratorii necesită ajutorul membrilor familiei, prietenilor sau unui pneumolog. Mulți oameni folosesc o vestă gonflabilă pentru piept care vibrează la o frecvență înaltă.

- Medicamente inhalatorii cu efecte bronhodilatatoare, drenante (mucolitice) și antibacteriene (de exemplu, fluorochinolone).
- Preparate care conțin enzime pancreatice pentru îmbunătățirea digestiei. Aceste preparate se iau în timpul meselor.
- Multivitamine (inclusiv vitamine liposolubile).
În 2015, FDA a aprobat un al doilea medicament pentru tratarea fibrozei chistice, care vizează o proteină defectă cunoscută sub numele de CFTR. Primul medicament, un așa-numit modulator CFTR, a fost aprobat în 2012. Se așteaptă ca modulatorii CFTR să prelungească viața unor persoane cu fibroză chistică cu decenii.
Intervenția chirurgicală poate fi necesară pentru a trata următoarele complicații respiratorii:
- Pneumotorax, hemoptizie masivă recurentă sau persistentă, polipi nazali, sinuzită persistentă și cronică.
- Ileus meconial, invaginație intestinală, prolaps rectal.
Transplantul pulmonar se efectuează în stadiul terminal al bolii.
Prognoză
Vârsta medie de supraviețuire a pacienților cu fibroză chistică variază între 35 și 40 de ani. Vârsta medie de supraviețuire este mai mare la bărbați decât la femei.
Cu strategii moderne de tratament, 80% dintre pacienți ajung la vârsta adultă. Cu toate acestea, fibroza chistică limitează semnificativ capacitățile funcționale ale pacientului. Încă nu există un leac pentru această boală.