Expert medical al articolului

Noile publicații
Boala Charcot-Marie-Tooth.
Ultima examinare: 12.07.2025

Tot conținutul iLive este revizuit din punct de vedere medical sau verificat pentru a vă asigura cât mai multă precizie de fapt.
Avem linii directoare de aprovizionare stricte și legătura numai cu site-uri cu reputație media, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale revizuite de experți. Rețineți că numerele din paranteze ([1], [2], etc.) sunt link-uri clickabile la aceste studii.
Dacă considerați că oricare dintre conținuturile noastre este inexactă, depășită sau îndoielnică, selectați-o și apăsați pe Ctrl + Enter.
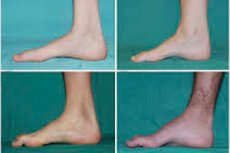
Atrofia musculară peronierală, sindromul sau boala Charcot-Marie-Tooth este un grup de boli ereditare cronice cu afectarea nervilor periferici.
Conform ICD-10, în secțiunea despre bolile sistemului nervos, codul pentru această boală este G60.0 (neuropatie motorie și senzorială ereditară). De asemenea, este inclusă în lista bolilor orfane.
Epidemiologie
Conform statisticilor clinice, prevalența tuturor tipurilor de boală Charcot-Marie-Tooth la 100 mii de locuitori este de 19 cazuri (conform altor surse, un caz la 2,5-10 mii de locuitori).
CMT tip 1 reprezintă aproximativ două treimi din cazuri (un caz la 5.000 până la 7.000 de locuitori), iar aproape 70% dintre acestea sunt asociate cu duplicarea genei PMP22. Peste 1,2 milioane de oameni din întreaga lume suferă de acest tip de boală.
Incidența CMT tip 4 este estimată la 1-5 cazuri la 10.000 de copii. [ 1 ]
Cauze Boala Charcot-Marie-Tooth
Conform clasificării sindroamelor polineuropatice, atrofia musculară peroneală (fibulară), amiotrofia neuronală Charcot-Marie-Tooth sau boala Charcot-Marie-Tooth (prescurtată CMT) se referă la polineuropatii motorii-senzoriale determinate genetic. [ 2 ]
Adică, cauzele apariției sale sunt mutațiile genetice. Și în funcție de natura deviațiilor genetice, se disting principalele tipuri sau forme ale acestui sindrom: demielinizante și axonale. Primul grup include boala Charcot-Marie-Tooth tip 1 (CMT1), care apare din cauza duplicării genei PMP22 pe cromozomul 17, care codifică proteina mielinică periferică transmembranară 22. Ca urmare, apare demielinizarea segmentară a tecii axonale (procesele celulelor nervoase) și o scădere a vitezei de conducere a semnalului nervos. În plus, pot exista mutații și în alte gene.
Forma axonală este boala Charcot-Marie-Tooth tip 2 (CMT2), care afectează axonii înșiși și este asociată cu modificări patologice ale genei MFN2 la locusul 1p36.22, care codifică proteina membranară mitofusin-2, necesară pentru fuziunea mitocondriilor și formarea rețelelor mitocondriale funcționale în interiorul celulelor nervoase periferice. Există peste o duzină de subtipuri de CMT2 (cu mutații în gene specifice).
Trebuie menționat că în prezent au fost identificate peste o sută de gene a căror afectare, transmisă prin moștenire, provoacă diverse subtipuri ale bolii Charcot-Marie-Tooth. De exemplu, mutațiile genei RAB7 duc la CMT tip 2B; modificarea genei SH3TC2 (care codifică una dintre proteinele membranare ale celulelor Schwann) provoacă CMT tip 4C, care se manifestă în copilărie și se caracterizează prin demielinizarea neuronilor motori și senzoriali (există o duzină și jumătate de forme de tip 4 ale acestei boli).
O formă rară de CMT de tip 3 (numită sindrom Dejerine-Sottas) începe să se dezvolte în copilăria timpurie și este cauzată de mutații ale genelor PMP22, MPZ, EGR2 și ale altor gene.
Când CMT tip 5 apare la vârsta de 5-12 ani, se observă nu numai neuropatie motorie (sub formă de parapareză spastică a extremităților inferioare), ci și leziuni ale nervilor optici și auditivi.
Slăbiciunea musculară și atrofia optică (cu pierderea vederii), precum și problemele de echilibru sunt tipice pentru CMT de tip 6. Iar în boala Charcot-Marie-Tooth de tip 7 există nu doar neuropatie motorie-senzorială, ci și o boală retiniană sub formă de retinită pigmentară.
Mai frecventă în rândul bărbaților, CMT legată de cromozomul X sau boala Charcot-Marie-Tooth cu tetrapareză a membrelor (slăbiciune a mișcării ambelor brațe și picioare) este un tip demielinizant și se crede că rezultă dintr-o mutație a genei GJB1 de pe brațul lung al cromozomului X, care codifică conexina 32, o proteină transmembranară a celulelor Schwann și a oligodendrocitelor ce reglează transmiterea semnalelor nervoase. [ 3 ]
Factori de risc
Principalul factor de risc pentru CMT este un istoric familial al bolii, adică la rude apropiate.
Conform geneticienilor, dacă ambii părinți sunt purtători ai genei autozomal recesive pentru boala Charcot-Marie-Tooth, riscul de a avea un copil care va dezvolta această boală este de 25%. Iar riscul ca acel copil să fie purtător al acestei gene (dar să nu prezinte niciun simptom) este estimat la 50%.
În cazul moștenirii legate de cromozomul X (când gena mutată se află pe cromozomul X al femeii), există un risc de 50% ca mama să transmită gena fiului ei, care va dezvolta CMT. Boala poate să nu apară atunci când se naște o fetiță, dar fiii (nepoții) fiicei pot moșteni gena defectă, iar boala se va dezvolta.
Patogeneza
În orice tip de boală Charcot-Marie-Tooth, patogeneza acesteia este cauzată de o anomalie ereditară a nervilor periferici: motori (de mișcare) și senzoriali (sensitivi).
Dacă tipul CMT este demielinizant, atunci distrugerea sau defectul tecii de mielină care protejează axonii nervilor periferici duce la o încetinire a transmiterii impulsurilor nervoase în sistemul nervos periferic - între creier, mușchi și organele senzoriale.
În tipul axonal al bolii, axonii înșiși sunt afectați, ceea ce afectează negativ puterea semnalelor nervoase, care este insuficientă pentru stimularea completă a mușchilor și a organelor senzoriale.
Citește și:
Cum se transmite sindromul Charcot-Marie-Tooth? Genele defecte pot fi moștenite autosomal dominant sau autosomal recesiv.
Cel mai frecvent tip, moștenirea autosomal dominantă, apare atunci când există o copie a genei mutate (purtată de unul dintre părinți). Iar probabilitatea transmiterii CMT fiecărui copil născut este estimată la 50%. [ 4 ]
În cazul moștenirii autozomal recesive, sunt necesare două copii ale genei defective (câte una de la fiecare părinte care nu prezintă semne ale bolii) pentru ca boala să se dezvolte.
În 40-50% din cazuri, apare demielinizarea ereditară autosomal dominantă, adică CMT tip 1; în 12-26% din cazuri, CMT axonală, adică tip 2. Iar în 10-15% din cazuri, se observă moștenire legată de cromozomul X. [ 5 ]
Simptome Boala Charcot-Marie-Tooth
De obicei, primele semne ale acestei boli încep să apară în copilărie și adolescență și se dezvoltă treptat pe parcursul vieții, deși sindromul se poate manifesta mai târziu. Combinația de simptome este variabilă, iar rata de progresie a bolii, precum și severitatea acesteia, sunt imposibil de prezis.
Simptomele tipice ale etapei inițiale includ oboseală generală crescută; scăderea tonusului (slăbiciune) a mușchilor picioarelor, gleznelor și tibiei; lipsa reflexelor. Acest lucru îngreunează mișcarea picioarelor și duce la disbazie (tulburări de mers) sub forma unei ridicări mai mari a picioarelor, adesea cu împiedicări și căderi frecvente. Semnele bolii Charcot-Marie-Tooth la un copil mic pot include stângăcie pronunțată și dificultăți de mers necaracteristice vârstei, asociate cu căderea bilaterală a piciorului. Deformările piciorului sunt, de asemenea, caracteristice: arcadă înaltă (picior scobit) sau picioare plate severe, degete curbate (în formă de ciocan).
În cazul mersului pe degetele de la picioare pe fondul hipotoniei musculare, un neurolog poate suspecta că un copil are CMT de tip 4, în care copiii pot fi incapabili să meargă până la adolescență.
Pe măsură ce boala progresează, atrofia musculară și slăbiciunea se extind la extremitățile superioare, îngreunând executarea abilităților motorii fine și efectuarea sarcinilor manuale normale. Scăderea senzațiilor tactile și a capacității de a simți căldura și frigul, precum și amorțeala la nivelul picioarelor și mâinilor, indică deteriorarea axonilor nervilor senzoriali.
În boala Charcot-Marie-Tooth tipurile 3 și 6, care se manifestă în copilărie, se observă ataxie senzorială (afectarea coordonării mișcărilor și a echilibrului), spasme și tremor muscular, leziuni ale nervului facial, atrofie a nervului optic cu nistagmus și pierderea auzului.
În stadiile ulterioare, pot apărea tremor (tremurături) incontrolabile și crampe musculare frecvente; problemele de mișcare pot duce la apariția durerii: musculare, articulare, neuropatice.
Complicații și consecințe
Boala Charcot-Marie-Tooth poate avea complicații și consecințe precum:
- entorse și fracturi mai frecvente;
- contracturi asociate cu scurtarea mușchilor și tendoanelor periarticulare;
- scolioză (curbura coloanei vertebrale);
- probleme respiratorii – atunci când fibrele nervoase care inervează mușchii diafragmei sunt deteriorate:
- pierderea capacității de a se mișca independent.
Diagnostice Boala Charcot-Marie-Tooth
Diagnosticul include examenul clinic, anamneza (inclusiv antecedentele familiale), examenul neurologic și sistemic.
Se efectuează teste pentru a verifica amplitudinea mișcărilor, sensibilitatea și reflexele tendinoase. Conducția nervoasă poate fi evaluată folosind diagnostice instrumentale - electromiografie sau electroneuromiografie. De asemenea, pot fi necesare ecografie sau RMN. [ 6 ]
Testarea genetică sau ADN pentru detectarea celor mai frecvente mutații genetice care cauzează CMT într-o probă de sânge este limitată, deoarece testele ADN nu sunt disponibile în prezent pentru toate tipurile de boli. Pentru mai multe informații, consultați Testarea genetică.
În unele cazuri, se efectuează o biopsie a nervului periferic (de obicei, nervul sural).
Diagnostic diferentiat
Diagnosticul diferențial include alte neuropatii periferice, distrofia musculară Duchenne, sindroamele mielopatice și miastenice, neuropatia diabetică, mielopatiile din scleroza laterală multiplă și amiotrofică, sindromul Guillain-Barré, traumatismele nervului peronier și atrofia acestuia (inclusiv la compresia între discurile lombare ale coloanei vertebrale), leziunile cerebelului sau talamusului, precum și efectele secundare ale chimioterapiei (în timpul tratamentului cu citostatice precum Vincristină sau Paclitaxel). [ 7 ]
Cine să contactați?
Tratament Boala Charcot-Marie-Tooth
Astăzi, tratamentul pentru această boală ereditară include terapia prin exerciții fizice (care vizează întărirea și întinderea mușchilor); terapia ocupațională (care ajută pacienții cu slăbiciune musculară la nivelul mâinilor); și utilizarea de dispozitive ortopedice pentru facilitarea mersului. Dacă este necesar, se prescriu analgezice sau anticonvulsivante. [ 8 ]
În cazurile de picioare plate severe, se poate efectua osteotomie, iar în cazul deformării călcâiului, este indicată corecția chirurgicală - artrodeză. [ 9 ]
Se desfășoară cercetări atât asupra componentei genetice a bolii, cât și asupra metodelor de tratament ale acesteia. Utilizarea celulelor stem, a anumitor hormoni, a lecitinei sau a acidului ascorbic nu a dat încă rezultate pozitive.
Însă, datorită cercetărilor recente, este posibil să apară într-adevăr ceva nou în tratamentul bolii Charcot-Marie-Tooth în viitorul apropiat. Astfel, din 2014, compania franceză Pharnext dezvoltă, iar de la mijlocul anului 2019, au loc studii clinice pentru medicamentul PXT3003 pentru tratamentul CMT de tip 1 la adulți, care suprimă expresia crescută a genei PMP22, îmbunătățește mielinizarea nervilor periferici și ameliorează simptomele neuromusculare.
Specialiștii de la compania medicală Sarepta Therapeutics (SUA) lucrează la crearea unei terapii genice pentru boala Charcot-Marie-Tooth de tip 1. Această terapie va utiliza un virus adeno-asociat (AAV) inofensiv din genul Dependovirus, cu un genom ADN monocatenar liniar, care va transfera în organism gena NTF3, care codifică proteina neurotrofină-3 (NT-3) necesară funcționării celulelor nervoase Schwann.
Helixmith va începe studiile clinice ale terapiei genice Engensis (VM202), dezvoltată în Coreea de Sud, până la sfârșitul anului 2020, pentru a trata simptomele musculare în CMT de tip 1. [ 10 ]
Profilaxie
Prevenirea CMT poate fi prin consiliere genetică a viitorilor părinți, mai ales dacă cineva din cuplu are antecedente familiale ale bolii. Cu toate acestea, au fost identificate cazuri de mutații genetice punctuale de novo, adică în absența bolii în antecedentele familiale.
În timpul sarcinii, probabilitatea apariției bolii Charcot-Marie-Tooth la viitorul copil poate fi verificată prin biopsie de vilozități coriale (de la 10 la 13 săptămâni de gestație), precum și prin analiza lichidului amniotic (la 15-18 săptămâni).
Prognoză
În general, prognosticul pentru diferite tipuri de boală Charcot-Marie-Tooth depinde de severitatea clinică, dar în toate cazurile boala progresează lent. Mulți pacienți au dizabilități, deși acest lucru nu reduce speranța de viață.