Expert medical al articolului

Noile publicații
Efectele ereditare (sindromul Alport) la copii
Ultima examinare: 23.04.2024

Tot conținutul iLive este revizuit din punct de vedere medical sau verificat pentru a vă asigura cât mai multă precizie de fapt.
Avem linii directoare de aprovizionare stricte și legătura numai cu site-uri cu reputație media, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale revizuite de experți. Rețineți că numerele din paranteze ([1], [2], etc.) sunt link-uri clickabile la aceste studii.
Dacă considerați că oricare dintre conținuturile noastre este inexactă, depășită sau îndoielnică, selectați-o și apăsați pe Ctrl + Enter.
Nefrita ereditara (sindromul Alport) - determinate genetic non-imune moștenit glomerulopathy hematurie expozante (uneori proteinurie), scăderea progresivă a funcției renale în dezvoltarea insuficienței renale cronice este adesea asociată cu surditate neurosenzorială și cu deficiențe de vedere.
Pentru prima dată boala a fost descrisă în 1902 de LGGuthrie, care a observat o familie din mai multe generații, din care sa observat hematurie. În 1915, membrii aceleiași familii AFHurst au descris dezvoltarea uremiei. În 1927, A Alport a identificat mai întâi surzenia în mai multe rude cu hematurie. În anii '50 ai secolului trecut, leziunile oculare au fost descrise într-o astfel de boală. În 1972, la pacienții cu hematurie ereditară, examinând morfologic țesutul renal, Hinglais et al. A arătat o expansiune inegală și o delaminare a membranelor bazale glomerulare. În 1985, sa identificat baza genetică a nefritei ereditare - o mutație în genele colagenului tip IV (Fiengold et al., 1985).
Analiza naturii genetice a bolii a făcut posibilă concluzia că diferențele manifestărilor fenotipice ale nefritei ereditare (cu sau fără pierderea auzului) se datorează gradului de exprimare a genei mutante. Astfel, în prezent, toate variantele clinice sunt considerate ca manifestări ale unei singure boli, iar termenul "nefrita ereditară" este sinonim cu termenul "sindrom Alport".
Conform studiilor epidemiologice, nefrită ereditară apare la o frecvență de 17 la 100.000 de copii.
Cauzele sindromului Alport
Baza genetică a bolii este o mutație în gena a-5 a lanțului de colagen de tip IV. Acest tip de universal pentru membranele bazale de rinichi, dispozitiv cohlear, capsule, lentile retiniene și cornee, care este prezentat în studiile folosind anticorpi monoclonali împotriva fracțiunii de colagen. Recent, ele indică posibilitatea utilizării probelor ADN pentru diagnosticarea prenatală a nefritei ereditare.
Se pune accentul pe importanța testării tuturor membrilor familiei utilizând sonde ADN pentru a identifica purtătorii genei mutante, ceea ce are o importanță deosebită în efectuarea consilierii medicale genetice a familiilor cu această boală. Cu toate acestea, până la 20% din familii nu au rude cu boală renală, ceea ce sugerează o incidență ridicată a mutațiilor spontane în gena anormală. Majoritatea pacienților cu nefrită ereditară în familie au indivizi cu boală renală, pierderea auzului și patologia vederii; legate de căsătorie între persoane care au unul sau mai mulți strămoși, deoarece căsătoria persoanelor înrudite sporește probabilitatea obținerii acelorași gene de la ambii părinți. Se stabilește autozomal dominant și autosomal recesiv și dominant, legat de cromozomul X al căii de transmisie.
Copiii sunt mai predispusi sa distinga trei variante de nefrita ereditara: sindromul Alport, nefrita ereditara fara pierderea auzului si hematuria familiala benigna.
Sindromul Alport - nefrită ereditară cu afectarea auzului. Baza este un defect combinat în structura colagenului membranei bazale a glomerulilor rinichilor, structurilor urechii și ochiului. Gena sindromului clasic Alport este localizată la locul 21-22 q al brațului lung al cromozomului X. În cele mai multe cazuri, este moștenit de tipul dominant, legat de cromozomul X. În acest sens, la bărbați, sindromul Alport este mai dificil, deoarece la femei funcția genei mutante este compensată de o alelă sănătoasă a celui de-al doilea cromozom intact.
Baza genetică a dezvoltării nefritei ereditare sunt mutații în genele lanțurilor alfa ale colagenului de tip IV. Este cunoscut ca șase lanțuri de colagen tip IV G: lanțuri de gene A5 si A6 (Sol4A5 și Sol4A5) sunt situate pe brațul lung al cromozomului X în zona 21-22q; genele de lanțuri a3 și a4 (Co4A3 și Co4A4) - pe cel de-al doilea cromozom; genele lanțurilor a1 și a2 (Co4A1 și Co4A2) - pe cromozomul 13.
În cele mai multe cazuri (80-85%), un tip de moștenire a bolii asociat cu X este asociat cu deteriorarea genei Co4A5 din cauza ștergerii, mutațiilor punctuale sau a tulburărilor de îmbinare. În prezent, mai mult de 200 de mutații ale genei Kol4A5, responsabile de încălcarea sintezei de lanțuri a5 de tip IV de colagen, se găsesc. În acest tip de moștenire, boala se manifestă la copii de ambele sexe, dar la băieți este mai dificilă.
Mutațiile în locurile genelor Co4A3 și Co4A4, responsabile de sinteza lanțurilor a3 și a4 ale colagenului IV, sunt moștenite autosomal. Conform cercetării, tipul dominant autosomal de moștenire este observat în 16% din cazurile de nefrită ereditară, autosomal recesiv - la 6% dintre pacienți. Există aproximativ 10 mutații ale genelor Co4A3 și Co4A4.
Rezultatul mutațiilor este o încălcare a proceselor de asamblare a colagenului de tip IV, ceea ce duce la o întrerupere a structurii acestuia. Colagenul de tip IV este una dintre principalele componente ale membranei bazale glomerulare, aparatul cohlear și lentila ochiului, care este detectată patologie în clinica nefrita ereditară.
Colagenul de tip IV, parte a membranei bazale glomerulare, constă în principal din două lanțuri a1 (IV) și un lanț a2 (IV), și conține, de asemenea, a3, a4, a5 lanț. Cel mai adesea atunci când moștenirea Sol4A5 mutatie X-linked însoțită de a3 lipsă, A4- și lanțurile A5 a6 de colagen de tip IV în structura și numărul de O1 și lanțurile a2 în glomerulare crește membranei bazale. Mecanismul acestui fenomen este neclar, se presupune că cauza este modificările posttranscripționale în ARNm.
A3 Lipsa, A4- și lanțuri a5 în structura de tip membrana IV de colagen subsol rezultatelor glomerulii subțierea și fragilitatea stadiile timpurii ale sindromului Alport care se manifestă majoritatea hematurie (uneori hematurie sau proteinurie numai proteinurie) punct de vedere clinic, pierdere și lenticonus auzului. Progresia ulterioară a bolii duce la îngroșarea și perturbarea permeabilitatea membranei bazale în stadiile avansate ale bolii, odată cu creșterea numărului acestor tipuri de colagen V și VI, manifestate în creșterea proteinuriei și reducerea funcției renale.
Natura mutației care stă la baza nefritei ereditare determină în mare măsură manifestarea fenotipică a acesteia. Cand X deleții cromozomiale cu mutație simultană și genele Sol4A6 Sol4A5 responsabile pentru sinteza lanțurilor A5 si A6 de colagen de tip IV, combinat cu sindromul Alport leiomyomatosis esofag și organele genitale. Conform studiilor cu mutații ale genei Sol4A5 asociate cu o deleție sunt marcate severitate mare a procesului patologic, o combinație cu o leziune renală manifestări extrarenale și dezvoltarea timpurie a insuficienței renale cronice, comparativ cu mutația stochechnoy a acestei gene.
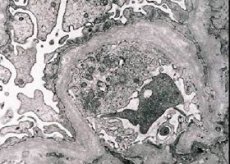
Din punct de vedere morfologic, microscopia electronică evidențiază subțierea și delaminarea membranelor bazale glomerulare (în special lamina densa) și prezența granulelor densă electronic. Leziunea glomerulului poate fi neuniformă la același pacient, de la leziunea focală minimă a mesangiului la glomeruloscleroza. Glomerulita în sindromul Alport este întotdeauna imun-negativă, ceea ce o diferențiază de glomerulonefrită. Caracteristică sunt dezvoltarea atrofiei canalului, infiltrarea limfohistiocitelor, prezența "celulelor spumate" cu incluziuni de lipide - lipofagi. Odată cu progresia bolii, se dezvăluie o îngroșare și o distrugere marcată a membranelor bazale glomerulilor.
Se dezvăluie anumite modificări ale stării sistemului imunitar. Pacienții cu nefrita ereditară scăderea nivelului de Ig A, și o tendință la o creștere a concentrației sanguine a IgM, IgG nivel poate fi crescută la stadiile timpurii ale bolii și declinul în etapele ulterioare. Poate că o creștere a concentrației de IgM și G este un fel de răspuns compensator ca răspuns la un deficit de IgA.
Activitatea funcțională a sistemului limfocitelor T este redusă; Acesta a marcat reducerea selectivă a limfocitelor B, responsabile pentru sinteza Ig A, rupt legătură imunitate fagocitare, în principal din cauza defalcare chemotaxiei proceselor și digestia intracelulară a neutrofilelor
In studiul biopsie renală la pacienții cu sindrom Alport prin microscopie electronică, modificări ultrastructurale observate membranei bazale glomerulare: subțierea și membranelor bazale glomerulare Scindările violare cu schimbarea grosimii sale și contururi inegale. În stadiile incipiente ale nefritei ereditare, defectul determină subțierea și fragilitatea membranelor bazale glomerulare.
Diluarea membranelor glomerulare este un semn mai favorabil și este mai frecventă la fete. O caracteristică microscopică electronică mai constanta în nefrita ereditară este scindarea membranei bazale, iar gravitatea distrugerii acesteia se corelează cu severitatea procesului.

Simptomele sindromului Alport la copii
Primele simptome ale sindromului Alport sub formă de sindrom urinar izolat sunt mai frecvent detectate la copiii din primii trei ani de viață. În cele mai multe cazuri, boala este detectată accidental. Sindromul urinar este dezvăluit în timpul examinării preventive a copilului, înainte de a intra în instituția pentru copii sau în timpul ARVI. În cazul apariției patologiei în urină în timpul ARVI. În cazul nefritei ereditare, spre deosebire de glomerulonefrita dobândită, nu există o perioadă latentă.
În stadiul inițial al bolii, bunăstarea copilului suferă puțin, trăsătura caracteristică este persistența și persistența sindromului urinar. Unul dintre principalele semne este hematuria în grade diferite, observată în 100% din cazuri. Creșterea gradului de hematurie este observată în timpul sau după infecții ale tractului respirator, efort fizic sau după vaccinări preventive. Proteinuria, în majoritatea cazurilor, nu depășește 1 g / zi, la debutul bolii poate fi instabilă, deoarece procesul progresează proteinurie. Periodic, sedimentul urinar poate avea leucocitriu, cu o predominanță de limfocite, care este asociat cu dezvoltarea schimbărilor interstițiale.
În continuare există o încălcare a deteriorării parțiale a funcției renale a stării generale a pacientului: există intoxicație, slăbiciune musculară, hipotensiune arterială, de multe ori pierdere (în special băieți) viziune, uneori neclară auzului. Intoxicarea se manifestă ca paloare, oboseală, dureri de cap. În stadiul inițial al bolii, pierderea auzului în majoritatea cazurilor este detectată doar prin audiografie. Pierderea auzului în sindromul Alport poate să apară în diferite perioade ale copilăriei, dar cel mai adesea pierderea auzului este diagnosticată la vârsta de 6-10 ani. Pierderea auzului la copii începe la frecvențe înalte, atingând un grad considerabil de conducere a aerului și osului, trecând de la conducerea sonoră la surzenia care recepționează sunetul. Pierderea auzului poate fi unul dintre primele simptome ale bolii și poate preceda sindromul urinar.
În 20% din cazuri, pacienții cu sindrom Alport au modificări ale ochilor. Cel mai frecvent detectat anomalie de a cristalinului: sferofokiya, lenticonus din față, din spate sau mixt, o varietate de cataractă. În familiile cu sindrom Alport există o incidență semnificativă de miopie. Un număr de cercetători sunt în mod constant în aceste familii sărbători schimbări bilaterale perimakulyarnye ca granulații luminoase albicioase sau gălbui în corpul galben. Ei consideră că acest simptom este un simptom constant, care are o valoare diagnostică ridicată în sindromul Alport. C. S. Chugh și colab. (1993) pentru studiul oftalmologică a aratat pacientii cu sindrom Alport scăderea acuității vizuale în 66,7% din cazuri, transmite lenticonus - 37,8%, petele de pe retină - la 22,2%, cataracta - 20%, keratoconus - 6 , 7%.
La unii copii cu nefrită ereditară, în special în formarea insuficienței renale, se observă un decalaj semnificativ în dezvoltarea fizică. Deoarece progresia insuficienței renale dezvoltă hipertensiune arterială. La copii, este mai frecvent detectat în adolescență și în grupurile de vârstă mai înaintată.
Caracteristic este prezența la pacienții cu nefrită ereditară a diferitelor (mai mult de 5-7) stigme ale diseminogenezei țesutului conjunctiv. Printre țesutul conjunctiv al stigmatului la pacientii cu cele mai frecvente hipertelorism ochi, cerul gurii de mare, malocluzii, forma anormală a urechilor, curbura degetul mic pe mâinile sale, „decalaj sandalevidnaya“ pe picioare. Pentru nefrita ereditară se caracterizează același tip de dizibriogeneză stigmă din cadrul familiei, precum și frecvența ridicată a răspândirii lor în rândul rudelor probandilor, prin care se transmite boala.
În stadiile incipiente ale bolii au relevat o reducere izolată a funcției renale parțiale: transportul de aminoacizi, electroliți, funcții de concentrare Acidogeneza, modificări suplimentare sunt starea funcțională atât proximal și nefronului distal și au caracterul unor tulburări parțiale combinate. Reducerea filtrării glomerulare are loc mai târziu, mai des în perioada adolescentă. Pe măsură ce progresează nefritele ereditare, se dezvoltă anemia.
Astfel, pentru ereditar nefrită stadializarea caracterizată de boală: primul stadiu latent sau simptome clinice ascunse manifestate prin sindromul vezicii modificări minime se produce atunci procesul de decompensare gradual, cu o reducere a funcției renale cu simptome evidente clinice (intoxicație, astenie, intarzieri de dezvoltare, anemizatsiya). Simptomele clinice apar, de obicei, indiferent de stratificarea reacției inflamatorii.
Efectele ereditare se pot manifesta în diferite perioade de vârstă, care depind de acțiunea genei, care până la un anumit timp este într-o stare reprimată.
Clasificare
Există trei variante de nefrită ereditară
- I - se manifestă clinic prin nefrită cu hematurie, pierderea auzului și afectarea ochilor. Cursul nefritei este progresiv în dezvoltarea CRF. Tipul de moștenire este dominant, legat de cromozomul X. Din punct de vedere morfologic, există o perturbare a structurii membranei bazale, subțierea și scindarea acesteia.
- II - se manifestă clinic prin nefrită cu hematurie fără pierderea auzului. Cursul nefritei este progresiv cu dezvoltarea insuficienței renale cronice. Tipul de moștenire este dominant, legat de cromozomul X. Din punct de vedere morfologic, se evidențiază subțierea membranei bazale a capilarelor glomerulare (în special laminadensa).
- III opțiune - hematurie familială benignă. Cursul este favorabil, insuficiența renală cronică nu se dezvoltă. Tipul de moștenire este autosomal dominant sau autosomal recesiv. În tipul autosomal recesiv de moștenire, femeile au un curs mai sever al bolii.
Diagnosticul sindromului Alport
Sunt propuse următoarele criterii:
- prezența în fiecare familie a cel puțin doi pacienți cu nefropatie;
- hematuria ca simptom principal al nefropatiei în proband;
- cel puțin un membru al familiei are o pierdere a auzului;
- dezvoltarea insuficienței renale cronice într-o rudă și mai mult.
În diagnosticul de o varietate de boli ereditare și congenitale un loc important aparține unei abordări integrate de inspecție și mai presus de toate acordând o atenție la datele obținute în pregătirea pedigree copilului. Diagnosticul sindromului Alport considerat valabil în cazurile în care pacientul 3 din 4 caracteristici tipice: prezența în hematuria familiei și insuficiență renală cronică, prezența pacientului pierderii de auz neurosenzoriale, patologii de detectare in electroni semne microscopice Caracterizarea biopsie clivaj membranei bazale glomerulare cu o modificare a grosimii sale și contururi neuniforme.
Examinarea pacientului trebuie să includă metode genetice clinice de investigare; studiu direct al anamnezei bolii; examinarea generală a pacientului ținând cont de criteriile de diagnostic. Etapa de compensare patologie poate captura doar concentrandu-se pe astfel de sindroame ca având antecedente familiale, hipotensiune arterială, mai multe stigmate dizembriogeneza schimbă sindromul vezicii urinare. In estrarenalnyh decompensată poate provoca simptome precum intoxicația severă, astenie, retard anemizatsiya dezvoltare fizică manifestată și amplificarea cu scăderea progresivă a funcției renale. La majoritatea pacienților cu scăderea funcției renale, se observă o scădere a funcției acido-și aminogenezei; la 50% dintre pacienți se observă o scădere semnificativă a funcției secretorii a rinichilor; limitarea intervalului de fluctuații ale densității optice a urinei; o încălcare a ritmului de filtrare și apoi o scădere a filtrării glomerulare. Etapa insuficiență renală cronică este diagnosticată prin prezența la pacienți timp de 3-6 luni sau niveluri mai ridicate ale ureei în ser (mai mult de 0,35 g / l), filtrare glomerulară redusă de până la 25% din normal.
Diagnosticul diferențial al nefrita ereditară trebuie realizată în primul rând cu glomerulonefrita forma hematuric dobândită. A câștigat glomerulonefrita tot mai acută începând perioada de 2-3 saptamani dupa o infectie anterioara, caracteristici extrarenale, inclusiv hipertensiunea cu primele zile (în nefrita ereditară, invers, hipotensiune), scăderea ratei de filtrare glomerulară la debut, nici o încălcare a funcțiilor tubulare parțiale, în timp ce ca și în cazul ereditar ei sunt prezenți. Glomerulonefrita Dobândite are loc cu hematurie și proteinurie mai severă, cu VSH crescut. Modificările tipice ale membranei bazale glomerulare caracteristice nefritei ereditare sunt de importanță diagnostică.
Diagnosticul diferential al nefropatiei dismetabolici efectuat cu insuficiență renală cronică în familie a identificat boala renala monotype punct de vedere clinic, și poate varia de la pielonefrita nefropatiei la urolitiaza. Copiii au adesea plângeri de dureri abdominale și periodic cu urinare, în sedimentul de urină - oxalat.
Dacă bănuiți că o nefrită ereditară trebuie trimisă pacientul pentru a clarifica diagnosticul într-un departament specializat de nefrologie.
Ce trebuie să examinăm?
Cum să examinăm?
Ce teste sunt necesare?
Cine să contactați?
Tratamentul sindromului Alport
În regimul prevede o limitare a exercițiilor fizice mari, stați pe aerul proaspăt. Dieta este de înaltă calitate, cu un conținut suficient de proteine de înaltă calitate, grăsimi și carbohidrați, luând în considerare funcția rinichilor. O mare importanță este identificarea și reabilitarea focarelor cronice de infecție. Din medicamente, se utilizează ATP, cocarboxilază, piridoxină (până la 50 mg / zi), clorură de carnitină. Cursurile se desfășoară de 2-3 ori pe an. Când hematuria este prescrisă fitoterapie - urzică, urzică, cenușă de mure, șarpe.
În literatura străină și în cea internă există rapoarte privind tratamentul cu prednisolon și utilizarea citostaticelor. Cu toate acestea, efectul este dificil de judecat.
În cazul insuficienței renale cronice, se utilizează hemodializă și transplant renal.
Nu există metode de terapie specifică (patogenetică eficientă) a nefritei ereditare. Toate măsurile medicale vizează prevenirea și încetinirea reducerii funcțiilor renale.
Dieta trebuie să fie echilibrată și calorică ridicată, ținând cont de starea funcțională a rinichilor. În absența unor încălcări ale stării funcționale în nutriția copilului ar trebui să fie un conținut suficient de proteine, grăsimi și carbohidrați. În prezența semnelor de disfuncție renală, cantitatea de proteine, carbohidrații de calciu și fosfor trebuie să fie limitată, ceea ce întârzie dezvoltarea insuficienței renale cronice.
Stresul fizic ar trebui limitat, iar copiii sunt sfătuiți să se abțină de la a face sport.
Evitați contactul cu pacienții infecțioși, reducând riscul de a dezvolta infecții respiratorii acute. Este necesară igienizarea focarelor de infecție cronică. Vaccinările preventive pentru copiii cu nefrită ereditară nu se efectuează, vaccinarea este posibilă numai în funcție de indicațiile epidemiologice.
Terapia hormonală și imunosupresoare în nefrita ereditară este ineficientă. Există indicii privind un anumit efect pozitiv (o scădere a nivelului de proteinurie și o încetinire a progresiei bolii) cu utilizarea pe termen lung a ciclosporinei A și a inhibitorilor ACE timp de mulți ani.
În tratamentul pacienților care utilizează medicamente care îmbunătățesc metabolismul:
- piridoxină - 2-3 mg / kg / zi în 3 doze divizate timp de 4 săptămâni;
- kokarboksilaza - 50 mg intramuscular la fiecare zi, doar 10-15 injecții;
- ATP - 1 ml intramuscular la fiecare zi, 10-15 injecții;
- Vitamina A - 1000 U / an / zi în 1 recepție timp de 2 săptămâni;
- vitamina E - 1 mg / kg / zi în 1 recepție timp de 2 săptămâni.
O astfel de terapie îmbunătățește starea generală a pacienților, reduce disfuncția tubulară și se administrează de 3 ori pe an.
Ca imunomodulator se poate folosi levamizol - 2 mg / kg / zi de 2-3 ori pe săptămână, cu întreruperi între doze de 3-4 zile.
Pentru cercetători, oxigenarea hiperbară are un efect pozitiv asupra severității hematuriei și a disfuncției renale.
Cea mai eficientă metodă de tratare a nefritei ereditare este transplantul renal în timp util. Când acest lucru nu este observat în recidiva transplant, într-un procent mic (aproximativ 5%) poate nefrită dezvoltarea în rinichiul, asociat cu antigene din membrana bazală glomerulară.
O zonă promițătoare este diagnosticul prenatal și terapia de inginerie genetică. Experimentele pe animale prezintă o eficiență ridicată a transferării genelor normale responsabile de sinteza lanțurilor a unui colagen de tip IV la țesutul renal, după care se observă sinteza structurilor normale de colagen.
Perspectivă
Prognosticul nefrită ereditară este întotdeauna gravă.
Criteriile defavorabile din punct de vedere al prognosticului pentru fluxul nefritei ereditare sunt:
- sex masculin;
- dezvoltarea precoce a insuficienței renale cronice la membrii familiei;
- proteinurie (mai mult de 1 g / zi);
- îngroșarea membranelor bazale glomerulare în funcție de microscopie;
- nevrită a nervului auditiv;
- deleție în gena Co4A5.
Prognosticul hematuriei familiale benigne este mai favorabil.
Использованная литература