Expert medical al articolului

Noile publicații
Prioni - agenți cauzali ai bolilor prionice
Ultima examinare: 06.07.2025

Tot conținutul iLive este revizuit din punct de vedere medical sau verificat pentru a vă asigura cât mai multă precizie de fapt.
Avem linii directoare de aprovizionare stricte și legătura numai cu site-uri cu reputație media, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale revizuite de experți. Rețineți că numerele din paranteze ([1], [2], etc.) sunt link-uri clickabile la aceste studii.
Dacă considerați că oricare dintre conținuturile noastre este inexactă, depășită sau îndoielnică, selectați-o și apăsați pe Ctrl + Enter.
Infecțiile virale lente sunt caracterizate prin criterii speciale:
- o perioadă de incubație neobișnuit de lungă (luni, ani);
- o leziune specifică a organelor și țesuturilor, în principal a sistemului nervos central;
- progresie lentă și constantă a bolii;
- rezultat fatal inevitabil.

Unii agenți patogeni care provoacă infecții virale acute pot provoca, de asemenea, infecții virale lente. De exemplu, virusul rujeolei provoacă uneori SSPE, iar virusul rubeolei provoacă rubeolă congenitală progresivă și panencefalită rubeolică.
O infecție virală lentă tipică la animale este cauzată de virusul Visna/Madi, care este un retrovirus. Acesta este agentul cauzal al infecției virale lente și al pneumoniei progresive la ovine. Substanța albă a creierului este distrusă, se dezvoltă paralizie (Visna - atrofiere); apare inflamația cronică a plămânilor și a splinei.
Bolile similare ca trăsături cu infecțiile virale lente sunt cauzate de prioni - agenții cauzali ai infecțiilor prionice. Bolile prionice sunt un grup de afecțiuni progresive ale sistemului nervos central la oameni și animale. La oameni, funcția sistemului nervos central este afectată, apar modificări de personalitate și tulburări de mișcare. Simptomele bolii durează de obicei de la câteva luni la câțiva ani, terminând cu deces. Anterior, infecțiile prionice erau considerate împreună cu așa-numiții agenți cauzali ai infecțiilor virale lente.
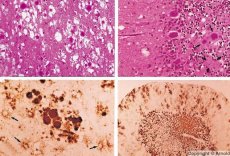
Unii agenți care cauzează bolile prionice se acumulează mai întâi în țesuturile limfoide. Prionii, la pătrunderea în creier, se acumulează în cantități mari, provocând amiloidoză (disproteinoză extracelulară, caracterizată prin depunerea de amiloid cu dezvoltarea atrofiei și sclerozei țesutului) și astrocitoză (proliferarea neurogliei astrocitare, hiperproducția de fibre gliale). Se formează fibrile, agregate de proteine sau amiloid și modificări spongiforme în creier (encefalopatii spongiforme transmisibile). Ca urmare, se produc modificări de comportament, coordonarea mișcărilor este afectată, se dezvoltă epuizare cu rezultat fatal. Nu se formează imunitate. Bolile prionice sunt boli conformaționale care se dezvoltă ca urmare a plierii incorecte (încălcării conformației corecte) a proteinelor celulare necesare funcționării normale a organismului. Căile de transmitere a prionilor sunt variate:
- cale alimentară - produse infectate de origine animală, aditivi alimentari din organe bovine crude etc.:
- transmitere prin transfuzie de sânge, administrarea de medicamente de origine animală, transplant de organe și țesuturi, utilizarea de instrumente chirurgicale și stomatologice infectate;
- transmiterea prin preparate imunobiologice (se cunoaște infectarea a 1500 de oi cu PrP''' prin vaccin cerebral cu formol provenit de la oi bolnave).
Prionii patologici, după ce au pătruns în intestin, sunt transportați în sânge și limfă. După replicarea periferică în splină, apendice, amigdale și alte țesuturi limfoide, aceștia sunt transferați în creier prin nervii periferici (neuroinvazie). Pătrunderea directă a prionilor în creier prin bariera hematoencefalică este posibilă. Anterior, se credea că sistemul nervos central este singurul țesut în care se acumulează prionii patologici, dar au apărut studii care au schimbat această ipoteză. S-a dovedit că acumularea de prioni în splină este asociată cu creșterea și funcționarea celulelor dendritice foliculare.
 [
[ Proprietățile prionilor
Izoforma celulară normală a proteinei prionice, cu o greutate moleculară de 33-35 kDa, este determinată de gena proteinei prionice (gena prionică - PrNP este situată pe al 20-lea cromozom uman). Gena normală apare la suprafața celulei (ancorată în membrană de glicoproteina moleculei), fiind sensibilă la protează. Aceasta reglează transmiterea impulsurilor nervoase, ciclurile zilnice, procesele de oxidare, participă la metabolismul cuprului în sistemul nervos central și la reglarea diviziunii celulelor stem din măduva osoasă. În plus, gena prionică se găsește în splină, ganglionii limfatici, piele, tractul gastrointestinal și celulele dendritice foliculare.
Proliferarea prionilor patologici
Transformarea prionilor în forme alterate are loc atunci când echilibrul controlat cinetic dintre aceștia este perturbat. Procesul este amplificat de o creștere a cantității de prion patologic (PrP) sau exogen. PrP este o proteină normală ancorată în membrana celulară. PrP' este o proteină globulară hidrofobă care formează agregate cu ea însăși și cu PrP'' pe suprafața celulei: ca urmare, PrP' se transformă în PrP'' și apoi ciclul continuă. Forma patologică a PrP''' se acumulează în neuroni, dând celulei un aspect spongios.
Kuru
Boala prionică, anterior frecventă printre papuani (care înseamnă tremur sau scuturare) în partea de est a insulei Noua Guinee. Proprietățile infecțioase ale bolii au fost dovedite de K. Gajdusek. Agentul patogen se transmite prin alimente ca urmare a canibalismului ritualic - consumul creierului insuficient gătit și infectat cu prioni al rudelor decedate. Ca urmare a deteriorării sistemului nervos central, mișcarea și mersul sunt afectate, apar frisoane și euforie („moartea prin râs”). Perioada de incubație durează 5-30 de ani. Pacientul decedează după un an.
Boala Creutzfeldt-Jakob
Boala prionică, care se manifestă ca demență, tulburări vizuale și cerebeloase și tulburări de mișcare cu rezultat fatal după 4-5 luni de boală în varianta clasică a bolii Creutzfeldt-Jakob și după 3-14 luni în noua variantă a bolii Creutzfeldt-Jakob. Perioada de incubație poate ajunge la 20 de ani. Sunt posibile diverse căi de infecție și cauze ale bolii:
- la consumul de produse animale insuficient tratate termic, cum ar fi carnea și creierul de la vaci cu encefalopatie spongiformă bovină;
- în timpul transplantului de țesuturi, cum ar fi transplantul de cornee, transfuzia de sânge, utilizarea hormonilor și a altor substanțe biologic active de origine animală, utilizarea catgutului, instrumente chirurgicale contaminate sau insuficient sterilizate, manipulări prosectoriale;
- în cazul hiperproducției de PrR și al altor afecțiuni care stimulează procesul de conversie a PrR' în PrR".
Boala se poate dezvolta și ca urmare a unei mutații sau inserții în regiunea genei prionice. Natura familială a bolii este frecventă datorită predispoziției genetice la boala Creutzfeldt-Jakob. În noua variantă a bolii Creutzfeldt-Jakob, tulburările se dezvoltă la o vârstă mai tânără (vârsta medie 28 de ani), spre deosebire de varianta clasică (vârsta medie 65 de ani). În noua variantă a bolii Creutzfeldt-Jakob, proteina prionică anormală se acumulează nu numai în sistemul nervos central, ci și în țesuturile limforeticulare, inclusiv în amigdale.
Sindromul Gerstmann-Sträussler-Scheinker
Boala prionică ereditară, însoțită de demență, hipotonie, tulburări de înghițire (disfagie), dizartrie. Adesea are o natură familială. Perioada de incubație este de la 5 la 30 de ani. Boala apare la 50-60 de ani, durata acesteia variind de la 5 la 13 ani.
Insomnie fatală ereditară
O boală autoimună cu insomnie progresivă, hiperreactivitate simpatică (hipertensiune arterială, hipertermie, hiperhidroză, tahicardie), tremor, ataxie, multiclonă, halucinații. Somnul este grav perturbat. Decesul survine odată cu progresia insuficienței cardiovasculare.
Racla
Scrapia (din engleza scrape - a răzui) este o boală prionică a oilor și caprelor (scabie), care apare cu afectarea sistemului nervos central, tulburări progresive de mișcare, mâncărime severă a pielii (scabie) și se termină cu moartea animalului.
Encefalopatia spongiformă bovină
O boală a bovinelor caracterizată prin afectarea sistemului nervos central, afectarea coordonării mișcărilor și moartea inevitabilă a animalului. Epidemia bolii a izbucnit pentru prima dată în Marea Britanie. A fost asociată cu hrănirea animalelor cu făină de carne și oase care conținea prioni patologici. Perioada de incubație variază de la 1,5 la 15 ani. Creierul, măduva spinării și globii oculari ai animalelor sunt cele mai infectate.
Diagnosticul de laborator al bolilor prionice
În timpul diagnosticării, se observă modificări spongiforme la nivelul creierului, astrocitoză (glioză) și absența infiltratelor inflamatorii. Creierul este colorat pentru amiloid. Markerii proteici ai tulburărilor cerebrale prionice sunt detectați în lichidul cefalorahidian (folosind ELISA). Se efectuează analiza genetică a genei prionice (PCR).
Prevenirea bolilor prionice
Pentru decontaminarea instrumentelor și a obiectelor din mediu se recomandă autoclavizarea (la 134°C timp de 18 min; la 121°C timp de 1 oră), incinerarea, tratamentul suplimentar cu înălbitor și o soluție normală de NaCl timp de 1 oră. Pentru profilaxia nespecifică, au fost introduse restricții privind utilizarea medicamentelor de origine animală și este interzisă producerea de hormoni hipofizari de origine animală. Transplantul de dura mater este restricționat. Se utilizează mănuși de cauciuc atunci când se lucrează cu fluidele de dialog ale pacienților.