Expert medical al articolului

Noile publicații
Mucopolizaharidoza tip 3
Ultima examinare: 23.04.2024

Tot conținutul iLive este revizuit din punct de vedere medical sau verificat pentru a vă asigura cât mai multă precizie de fapt.
Avem linii directoare de aprovizionare stricte și legătura numai cu site-uri cu reputație media, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale revizuite de experți. Rețineți că numerele din paranteze ([1], [2], etc.) sunt link-uri clickabile la aceste studii.
Dacă considerați că oricare dintre conținuturile noastre este inexactă, depășită sau îndoielnică, selectați-o și apăsați pe Ctrl + Enter.
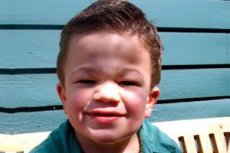
Mucopolysaccharidosis tip III (sinonime: sindromul Sanfilippo, insuficiență lizozomale aN-acetylglucosaminidase - mucopolysaccharidosis Sh A, acetil-CoA și-glucosaminidă-N-acetiltransferaza - mucopolysaccharidosis III B, N-acetilglucozamină-6-sulfatază - mucopolysaccharidosis III C, sulfamidazy - mucopolizaharidoză III D).
 [
[Patogeneza
Prin boala provoaca mutatii patru gene diferite: lizozomale aN-acetylglucosaminidase (mucopolysaccharidosis III A), acetil-CoA și-glucosaminidă-N-acetiltransferaza (mucopolysaccharidosis III B), lizozomale N-acetilglucozamină-6-sulfatază (mucopolysaccharidosis III C) sulfamidazy (mucopolizaharidoza III D). Toate enzimele sunt implicate în metabolizarea sulfatului de heparan.
Gena heparan-N-sulfatază-SGSH-este localizată pe brațul lung al cromozomului 17-17q25.3. 75,3% din mutațiile actuale cunoscute în gena SGSH sunt mutații punctuale. Au fost descrise mutații frecvente tipice pentru populațiile europene - R74C (56% în Polonia și 21% în Germania) și R245H (56% în Țările de Jos).
Frecvența mutației R74C este de 47,5%, mutația R245H este de 7,5%. Celelalte două mutații, delll35G și N389S, reprezintă împreună 21,7% din alelele mutante.
Gena aN-acetil-glucosaminidază, NAGLU, este localizată pe brațul lung al cromozomului 17-17q21. 69% din mutațiile găsite în gena NAGLU sunt mutații de tip missense și nonsens, 26,3% sunt deleții și inserții mici. Gena acetil-CoA-cc-glucosaminide-N-acetiltransferază, HGSNAT, este localizată pe brațul scurt al cromozomului 8-8p11. Gena a fost caracterizată doar în 2006 și, până în prezent, a găsit doar câteva mutații.
Gena N-acetil-glucozamină-6-sulfatază - GNS - este localizată pe brațul lung al cromozomului 12-12ql4. Există 12 pacienți cu mucopolizaharidoză IIID înregistrați în lume. Există 4 mutații în gena GNS.
Când toate subtipurile III încălcare mucopolysaccharidosis are loc o degradare a heparan sulfat, incluse în structura membranelor celulare, incluzând membranele de neuroni, care se corelează cu procesele neurodegenerative severe cauzate de atrofie corticală. Diareea cronică se explică prin implicarea în procesul patologic al sistemului nervos autonom, împreună cu disfuncția mucoasei intestinale. Pierderea senzorinurală a auzului se datorează, probabil, trei cauze: otita frecventă, deformările osiciilor auditive și anomaliile urechii interne. Rigiditatea articulațiilor este rezultatul deformării metaforelor, îngroșarea capsulei comune este secundară depunerii glicozaminoglicanilor și fibrozei în ea. Diferențele intrasinndomice în severitatea bolii se datorează exclusiv activității funcționale reziduale a enzimei mutante: cu cât este mai mare, cu atât mai ușoară progresează boala.
Simptome mucopolizaharidoza tip 3
Polimorfismul clinic în sindromul Sanfilippo este mai puțin pronunțat decât în cazul altor tipuri de mucopolizaharidoză. Caracterizat printr-o progresie lentă a bolii, tulburări neurologice severe cu simptome severe de la organele interne și alte sisteme.
Primele simptome ale bolii apar de obicei la vârsta de 2-6 ani la copii cu o dezvoltare normală anterioară. Simptomele manifestării includ regresia dezvoltării psihomotorii și a discursului, tulburările psihiatrice sub forma dezvoltării sindromului de hiperactivitate, comportamentul autistic sau agresiv, tulburările de somn; copiii devin neglijenți și neatenți.
Alte simptome comune sunt hirsutismul, părul dur, hepatosplenomegalia ușoară, deformitatea valgusului la nivelul extremităților, gâtul scurt. Formarea de caracteristici faciale grosiere de tip gargoilizma și deformare osoase multiple dysostosis slab exprimat la Mucopolysaccharidosis III, comparativ cu alte tipuri de mucopolysaccharidosis, caracterizat fenotip Hurler. Creșterea, de regulă, corespunde vârstei, iar rigiditatea articulațiilor rar provoacă o încălcare a funcțiilor lor. Majoritatea pacienților dezvoltă adesea osteoporoză și osteomalacie. Tulburările scheletale secundare sunt un risc ridicat de fracturi patologice. Tulburările psihonostomice severe sunt observate cel mai adesea în 6-10 ani de viață, duc la o maladie socială severă. Pierderea progresivă a auzului senzorinural este inerentă la toți pacienții cu forme severe și moderate ale bolii. Convulsiile sunt observate la aproape toți pacienții pe măsură ce progresează boala.
Cursul bolii progresează rapid, majoritatea pacienților nu supraviețuiesc până la vârsta de 20 de ani. Se crede că mucopolizaharidoza IIIA este tipul cel mai comun și sever al acestui sindrom.
Diagnostice mucopolizaharidoza tip 3
Diagnosticul mucopolizaharidoză III este confirmat prin determinarea nivelului de excreție și măsurarea activității enzimatice a glucosamicinei urinare. În cazul mucopolizaharidoză III, excreția totală a glicozaminoglicanilor în urină crește și se observă hiperexcreția heparanului sulfat. Activitatea enzimelor lizozomale care corespund unui anumit subtip de mucopolizaharidoză III este măsurată în leucocite sau o cultură a fibroblastelor cutanate utilizând un substrat fluorogen artificial.
Diagnosticul prenatal este posibilă prin măsurarea activității enzimei în chorionic villus biopsie la 9-11 săptămâni de gestație și / sau determinarea spectrului de glicozaminoglicani în lichidul amniotic la 20-22 săptămâni de sarcină. Pentru familiile cu un genotip cunoscut, este posibilă efectuarea diagnosticării ADN în stadiile incipiente ale sarcinii.
Ce teste sunt necesare?
Diagnostic diferentiat
Diagnosticul diferential se realizează în cadrul grupului Mucopolysaccharidosis, precum și cu alte tulburări lizozomale de stocare: Mucolipidosis, galaktosialidozom, sialidosis, mannozidozom, fucosidosis, GM1-gangliosidosis.
Cine să contactați?
Tratament mucopolizaharidoza tip 3
Până în prezent, terapiile eficiente pentru mucopolizaharidoza III nu au fost dezvoltate. Este indicată terapia simptomatică.
Использованная литература