Expert medical al articolului

Noile publicații
Sindromul Usher
Ultima examinare: 04.07.2025

Tot conținutul iLive este revizuit din punct de vedere medical sau verificat pentru a vă asigura cât mai multă precizie de fapt.
Avem linii directoare de aprovizionare stricte și legătura numai cu site-uri cu reputație media, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale revizuite de experți. Rețineți că numerele din paranteze ([1], [2], etc.) sunt link-uri clickabile la aceste studii.
Dacă considerați că oricare dintre conținuturile noastre este inexactă, depășită sau îndoielnică, selectați-o și apăsați pe Ctrl + Enter.
Sindromul Usher este o boală ereditară care se manifestă prin surditate completă de la naștere, precum și prin orbire progresivă odată cu înaintarea în vârstă. Pierderea vederii este asociată cu retinita pigmentară, un proces de degenerare pigmentară a retinei. Multe persoane cu sindrom Usher au, de asemenea, probleme severe de echilibru.
Epidemiologie
Datorită cercetărilor, s-a putut stabili că sindromul Usher afectează aproximativ 8% dintre copiii surdo-muți examinați (testele au fost efectuate în instituții speciale pentru persoane surdo-mute). Retinita pigmentară a fost observată la 6-10% dintre pacienții care suferă de surditate congenitală, care, la rândul ei, se observă la aproximativ 30% dintre persoanele cu boli pigmentare ale retinei.
Se crede că această boală se manifestă la aproximativ 3-10 persoane din 100 de mii la nivel mondial. Poate fi observată atât la femei, cât și la bărbați în mod egal. Aproximativ 5-6% din populația lumii suferă de acest sindrom. Aproximativ 10% din toate cazurile de surditate profundă la copii apar din cauza sindromului Usher I, precum și a tipurilor II.
În Statele Unite, tipurile 1 și 2 sunt cele mai frecvente. Împreună, acestea reprezintă aproximativ 90 până la 95% din toate cazurile de sindrom Usher la copii.
Cauze Sindromul Usher
Tipurile I, II și III ale sindromului Usher au o cauză autosomal recesivă, în timp ce tipul IV este considerat o tulburare a cromozomului X. Cauzele orbirii și surdității care apar în cazul acestui sindrom nu au fost încă suficient studiate. Se presupune că persoanele cu această boală sunt hipersensibile la componentele care pot deteriora structura ADN-ului. În plus, această boală poate fi asociată cu tulburări ale sistemului imunitar, dar în acest caz nu există o imagine exactă a acestui proces.
În 1989, anomaliile cromozomiale au fost identificate pentru prima dată la pacienții cu boală de tip II, ceea ce ar putea duce în viitor la o modalitate de izolare a genelor care cauzează sindromul. De asemenea, ar putea fi posibilă identificarea acestor gene la purtători și dezvoltarea unor teste genetice prenatale speciale.
 [ 8 ]
[ 8 ]
Factori de risc
Sindromul este moștenit atunci când ambii părinți sunt afectați, adică este moștenit de tip recesiv. Un copil poate moșteni boala și dacă părinții săi sunt purtători ai genei. Dacă ambii viitori părinți au această genă, atunci probabilitatea de a avea un copil cu acest sindrom este de 1 din 4. O persoană care are o singură genă pentru sindrom este considerată purtătoare, dar nu prezintă simptome ale tulburării. În zilele noastre, nu este încă posibil să se determine dacă o persoană are gena pentru această boală.
Dacă un copil se naște din părinți, dintre care unul nu are o astfel de genă, atunci probabilitatea ca acesta să moștenească sindromul este foarte mică, dar cu siguranță va fi purtător.
Simptome Sindromul Usher
Simptomele sindromului Usher includ pierderea auzului și acumularea anormală de celule pigmentate în structurile oculare. Pacientul dezvoltă apoi degenerare a retinei, ceea ce provoacă deteriorarea vederii și, în cele mai grave cazuri, pierderea vederii.
Pierderea auzului neurosenzorial poate fi ușoară sau completă și, de obicei, nu progresează de la naștere. Cu toate acestea, bolile pigmentare ale retinei pot începe să se dezvolte în copilărie sau mai târziu. Rezultatele testelor au arătat că acuitatea vizuală centrală poate fi menținută timp de mulți ani, chiar și atunci când vederea periferică se deteriorează (o afecțiune numită „vedere în tunel”).
Acestea sunt principalele manifestări ale bolii, care uneori pot fi completate de alte tulburări, cum ar fi psihoza și alte tulburări mintale, probleme cu urechea internă și/sau cataracta.
Formulare
În timpul cercetării, au fost identificate 3 tipuri ale acestei boli, precum și o a 4-a formă, care este destul de rară.
Tipul I al bolii este caracterizat prin surditate congenitală completă, precum și prin tulburări de echilibru. Adesea, acești copii încep să meargă abia la vârsta de 1,5 ani. Deteriorarea vederii începe de obicei la vârsta de 10 ani, iar dezvoltarea finală a stării de orbire nocturnă începe la vârsta de 20 de ani. Copiii cu acest tip de boală pot dezvolta o deteriorare progresivă a vederii periferice.
În boala de tip II se observă surditate moderată sau congenitală. În acest caz, deteriorarea surdității parțiale adesea nu mai apare. Retinita pigmentară începe să se dezvolte în jurul sfârșitului adolescenței sau după 20 de ani. Dezvoltarea orbirii nocturne începe de obicei la 29-31 de ani. Deficiența de acuitate vizuală în cazul patologiei de tip II progresează, în general, puțin mai lent decât în tipul I.
Tipul III al bolii este caracterizat prin pierderea progresivă a auzului, care începe de obicei în timpul pubertății, precum și prin dezvoltarea treptată în aceeași perioadă (puțin mai târziu decât pierderea auzului) a retinitei pigmentare, care poate deveni un factor în dezvoltarea orbirii progresive.
Manifestările patologiei de tip IV apar în principal la bărbați. În acest caz, se observă și tulburări progresive și pierderea auzului și a vederii. Această formă este foarte rară și are de obicei o natură cromozomială X.
Diagnostice Sindromul Usher
Diagnosticul sindromului Usher se stabilește pe baza combinației observate la pacient de surditate bruscă și pierdere progresivă a vederii.
Teste
Se poate comanda un test genetic special pentru a detecta mutația.
Au fost descoperite unsprezece loci genetici care pot cauza dezvoltarea sindromului Usher și au fost identificate nouă gene care sunt cu siguranță cauza tulburării:
- Tipul 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Tipul 2: ush2a, VLGR1, WHRN.
- Sindromul Usher tip 3: USH3A.
Oamenii de știință de la NIDCD, împreună cu colegi de la universități din New York și Israel, au identificat o mutație numită R245X în gena Pcdh15, care este responsabilă pentru un procent mare de sindrom Usher de tip 1 în populația evreiască.
Pentru a afla despre laboratoarele care efectuează studii clinice, vizitați https://www.genetests.org și căutați în directorul laboratoarelor „sindromul Usher”.
Pentru a afla despre studiile clinice existente care includ testarea genetică pentru sindromul Usher, vizitați https://www.clinicaltrials.gov și căutați „sindromul Usher” sau „testarea genetică a sindromului Usher”.
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Diagnosticare instrumentală
Există mai multe metode de diagnostic instrumental:
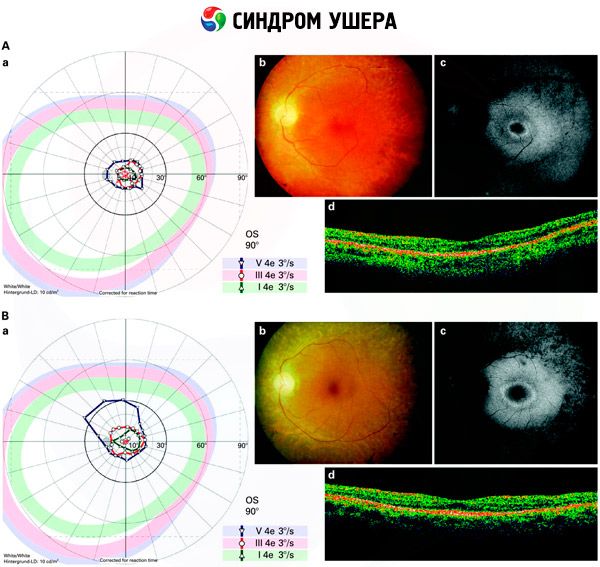
- Examinarea fundului de ochi pentru a detecta prezența petelor pigmentare pe retină, precum și îngustarea vaselor retiniene;
- Electroretinograma, care permite detectarea deviațiilor degenerative inițiale la nivelul retinei oculare. Aceasta arată dispariția căilor electroradiografice;
- O electronistagmogramă (ENG) măsoară mișcările involuntare ale ochilor care ar putea indica prezența unui dezechilibru.
- Audiometria, care este utilizată pentru a determina prezența surdității și severitatea acesteia.
Diagnostic diferentiat
Sindromul Usher trebuie diferențiat de unele tulburări similare.
Sindromul Hallgren, caracterizat prin pierderea congenitală a auzului și pierderea progresivă a vederii (se dezvoltă și cataractă și nistagmus). Alte simptome includ ataxie, tulburări psihomotorii, psihoză și retard mintal.
Sindromul Alstrom, o boală ereditară în care retina degenerează, ducând la pierderea vederii centrale. Acest sindrom este asociat cu obezitatea infantilă. În același timp, diabetul zaharat și pierderea auzului încep să se dezvolte după 10 ani.
Rubeola la o femeie însărcinată în primul trimestru de sarcină poate provoca diverse anomalii în dezvoltarea copilului. Printre consecințele unei astfel de anomalii se numără pierderea auzului, precum și (sau) probleme de vedere și, pe lângă acestea, diverse defecte de dezvoltare.
Cine să contactați?
Tratament Sindromul Usher
În prezent, nu există niciun leac pentru sindromul Usher. Prin urmare, terapia în acest caz constă în principal în încetinirea procesului de pierdere a vederii, precum și în compensarea pierderii auzului. Printre metodele de tratament posibile se numără:
- Administrarea de vitamina A (unii oftalmologi consideră că dozele mari de palmitat de vitamina A pot încetini, dar nu pot opri, progresia retinitei pigmentare);
- Implantarea de dispozitive electronice speciale în urechile pacientului (aparate auditive, implanturi cohleare).
Oftalmologii recomandă ca majoritatea adulților cu forme comune de retinită pigmentară să ia zilnic 15.000 UI (unități internaționale) de palmitat de vitamina A, sub supraveghere medicală. Deoarece persoanele cu sindrom Usher de tip 1 nu au fost incluse în studiu, dozele mari de vitamina A nu sunt recomandate pentru acest grup de pacienți. Persoanele care iau în considerare administrarea de vitamina A ar trebui să discute această opțiune de tratament cu medicul lor. Alte recomandări pentru această opțiune de tratament includ:
- Schimbarea dietei pentru a include alimente bogate în vitamina A.
- Femeile care intenționează să rămână însărcinate ar trebui să întrerupă administrarea de doze mari de vitamina A cu trei luni înainte de a concepe, din cauza unui risc crescut de malformații congenitale.
- Femeile însărcinate ar trebui să întrerupă administrarea de doze mari de vitamina A din cauza unui risc crescut de malformații congenitale.
De asemenea, este importantă adaptarea unui astfel de copil la viața socială. Acest lucru necesită ajutorul profesorilor de educație specială și al psihologilor. În cazul în care pacientul a început să experimenteze o pierdere progresivă a vederii, acesta ar trebui învățat să folosească limbajul semnelor.
Prognoză
Sindromul Usher are un prognostic nefavorabil. Câmpul vizual și acuitatea acestuia încep să se deterioreze în perioada de 20-30 de ani la majoritatea pacienților cu această boală, de orice tip. În unele cazuri, apare pierderea completă a vederii bilaterale. Pierderea auzului, care este întotdeauna însoțită de muțenie, evoluează foarte rapid până la o pierdere completă a auzului bilateral.