Expert medical al articolului

Noile publicații
Sindromul Angelman la copii și adulți
Last reviewed: 04.07.2025

Tot conținutul iLive este revizuit din punct de vedere medical sau verificat pentru a vă asigura cât mai multă precizie de fapt.
Avem linii directoare de aprovizionare stricte și legătura numai cu site-uri cu reputație media, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale revizuite de experți. Rețineți că numerele din paranteze ([1], [2], etc.) sunt link-uri clickabile la aceste studii.
Dacă considerați că oricare dintre conținuturile noastre este inexactă, depășită sau îndoielnică, selectați-o și apăsați pe Ctrl + Enter.

Există o serie de boli pentru care expresii precum „ai grijă de tine și nu te vei îmbolnăvi” sună, cel puțin, ridicole. Acestea sunt patologii în care unele anomalii mentale și fizice sunt inerente organismului copilului chiar înainte de naștere, dar părinții nu sunt de vină pentru acest lucru. Astfel de boli sunt cauzate de mutații sau anomalii ale seturilor de cromozomi și sunt numite cromozomiale sau genetice. Sindromul Angelman, sindromul Down, sindromul Patau, sindromul Edwards, sindromul Turner, sindromul Prader-Willi - aceasta este doar o parte a bolilor genetice dintr-o listă destul de decentă.
Sindromul omului fericit
De data aceasta vom vorbi despre patologia numită după medicul pediatru englez Harry Angelman, care a ridicat pentru prima dată problema acestei probleme în 1965, după ce cu o zi înainte întâlnise în cabinetul său trei copii neobișnuiți, uniți de simptome comune și specifice. Doctorul i-a numit pe acești copii „copii-păpuși” și a scris un articol despre ei, care inițial s-a intitulat „Copiii-marionete”. Articolul în sine și titlul său au fost scrise sub inspirația unui tablou văzut într-unul dintre muzeele din Verona. Tabloul înfățișa un băiat râzând și se numea „Băiatul-Păpușă”. Asocierea copilului înfățișat în tablou cu cei trei copii pe care Angelman i-a întâlnit cândva în cabinetul său l-a determinat pe medicul pediatru să-i combine pe copii într-un singur grup, din cauza bolii pe care o aveau.
Nu este nimic surprinzător în faptul că copiii menționați în articol nu au fost observați de alți medici. La urma urmei, la prima vedere părea că aveau boli complet diferite, atât de diferit era tabloul clinic general al bolii în 3 cazuri diferite. Poate că „noua” patologie cromozomială i-ar fi interesat pe alți oameni de știință, dar la acea vreme genetica nu era încă suficient dezvoltată pentru a confirma ipoteza medicului englez. Prin urmare, după un anumit interes pentru aceasta, articolul a fost aruncat pe raftul din spate pentru o lungă perioadă de timp.
Următoarea mențiune a sindromului Angelman, așa cum era numit acum articolul pediatrului englez G. Angelman, datează de la începutul anilor '80 ai secolului XX. Și abia în 1987 a fost posibil să se afle motivul pentru care o mică parte dintre copii se nasc cu astfel de abateri încât, din exterior, par să zâmbească constant și să fie fericiți. De fapt, acest lucru nu este deloc adevărat, iar zâmbetul este doar o grimasă, în spatele căreia se ascunde un suflet uman nefericit și durerea părinților.
Epidemiologie
Conform statisticilor, o mutație cromozomială la un copil se poate dezvolta atât pe fondul unor mutații similare la părinți, cât și în absența acestora. Nu există o natură ereditară clară a sindromului Angelman (AS), dar probabilitatea dezvoltării patologiei la părinții cu mutații cromozomiale este destul de mare.
De asemenea, este interesant faptul că, dacă o familie are deja un copil cu AS, există o șansă de unu la sută de a avea un al doilea copil cu aceeași tulburare, chiar dacă părinții sunt sănătoși.
Încă nu există statistici exacte privind numărul de pacienți cu sindrom Angelman. Poate că motivul este varietatea simptomelor, care pot apărea într-o anumită compoziție sau pot să nu apară deloc pentru o perioadă lungă de timp. Se presupune că prevalența bolii este: 1 copil la 20.000 de nou-născuți. Dar această cifră este foarte aproximativă.
Cauze Sindromul Angelman
Sindromul Angelman este o denumire medicală pentru o patologie cromozomială, dar este departe de a fi singura. Oamenii numesc această boală sindromul copiilor păpuși, sindromul păpușii fericite, sindromul Petrushka și sindromul păpușii râzătoare. Oamenii vin cu tot felul de nume (uneori chiar jignitoare pentru pacienții înșiși și pentru părinții lor), dar o boală este o boală, indiferent cât de amuzantă ar părea și indiferent de motive.
Și motivele dezvoltării sindromului Angelman, ca multe alte patologii genetice, sunt în toate cazurile tulburări ale structurii unuia dintre cromozomi sau a setului de cromozomi în ansamblu. Dar în cazul nostru, întreaga problemă constă în cromozomul 15, transmis de la mamă. Adică, cromozomul patern în acest caz nu are abateri, dar cel feminin suferă anumite mutații.
În funcție de tipul de anomalie cromozomială, sindromul Angelman este clasificat ca o mutație cromozomială. Astfel de mutații sunt considerate a fi:
- O deleție (absența unei secțiuni a unui cromozom care conține un anumit set de gene; dacă una dintre gene lipsește, vorbim despre o microdeleție), care este rezultatul a două rupturi și o reuniune, atunci când se pierde o secțiune a cromozomului original.
- Duplicarea (prezența unei secțiuni suplimentare într-un cromozom care este o copie a uneia existente), care în majoritatea cazurilor duce la moartea unei persoane și, mai rar, la infertilitate.
- Inversiune (inversarea uneia dintre secțiunile cromozomului cu 180 de grade, adică în direcția opusă, iar apoi genele din acesta sunt situate în ordinea opusă), când capetele rupte ale cromozomului sunt conectate într-o ordine diferită de cea originală.
- Inserție (dacă o parte a materialului genetic dintr-un cromozom este în afara locului),
- translocație (dacă o anumită secțiune a unui cromozom este atașată de un alt cromozom; o astfel de mutație poate fi reciprocă fără pierderea secțiunilor).
Primind un cromozom mutat de la o mamă neatentă, copilul este sortit să se nască cu anomalii. Cea mai frecventă cauză a sindromului Angelman este încă considerată a fi o deleție a celui de-al 15-lea cromozom matern, atunci când lipsește o mică secțiune. Mutațiile mai puțin frecvente în sindromul „păpușa râzătoare” sunt considerate a fi:
- translocație,
- disomie unipaternală (dacă copilul a primit o pereche de cromozomi de la tată, cromozomul matern este absent),
- mutația genelor din ADN, care sunt atât principalul material de construcție (genetic), cât și instrucțiuni pentru utilizarea corectă a acestuia (în special, mutația genei ube3a în cromozomul matern).
Prezența uneia dintre aceste mutații la părinți este un factor de risc pentru dezvoltarea sindromului Angelman la copii. Însă nu numai mutațiile cromozomiale, ci și cele genomice (care sunt asociate cu o modificare cantitativă a seturilor de cromozomi și sunt mai frecvente decât cele cromozomiale) pot provoca dezvoltarea bolii la un copil. Mutațiile genomice comune includ trisomia cromozomială (dacă setul de cromozomi al unei persoane are mai mult de 46 de cromozomi).
Pentru ca o patologie să apară la un copil, nu este deloc necesar ca părinții să prezinte anomalii cromozomiale. Și totuși, există un anumit procent de pacienți a căror boală este ereditară.
Patogeneza
Să aprofundăm puțin biologia, sau mai precis, genetica. Informația genetică a fiecărui organism uman este conținută în 23 de perechi de cromozomi. Un cromozom dintr-o pereche este transmis copilului de la tată, celălalt de la mamă. Toate perechile de cromozomi diferă ca formă și dimensiune și poartă anumite informații. Astfel, a 23-a pereche de cromozomi (cromozomii X și Y) este responsabilă pentru formarea caracteristicilor sexuale ale bebelușului (XX - fată, XY - băiat, în timp ce cromozomul Y poate fi primit de copil doar de la tată).
În mod ideal, un copil primește 46 de cromozomi de la părinți, care îi formează caracteristicile genetice, predeterminându-l ca individ. Un număr mai mare de cromozomi se numește trisomie și este considerat o abatere de la normă. De exemplu, prezența cromozomului 47 în setul de cromozomi (cariotip, determinarea speciei și a caracteristicilor individuale) provoacă apariția sindromului Down.
Dacă cromozomii sunt colorați cu un colorant special, atunci la microscop puteți vedea dungi de diferite nuanțe de-a lungul fiecăruia dintre ei. În interiorul fiecărei dungi există un număr imens de gene. Toate aceste dungi sunt numerotate de oamenii de știință și au o locație fixă. Absența uneia dintre dungi este considerată o abatere de la normă. În sindromul Angelman, se poate observa foarte des absența segmentelor cromozomului matern în intervalul q11-q13, situate în brațul lung, numărul de baze ADN în care este de doar aproximativ 4 milioane.
Componenta principală a cromozomului este considerată a fi o moleculă de ADN incredibil de lungă, care conține mii de gene și zeci și sute de milioane de baze azotate. Astfel, cromozomul 15, responsabil pentru dezvoltarea sindromului Angelman și a altora, conține 1200 de gene și aproximativ 100 de milioane de baze. Orice perturbare a structurii moleculei de ADN va afecta cu siguranță aspectul și dezvoltarea viitorului copil.
Informația genetică conținută în gene este convertită în proteine sau ARN. Acest proces se numește expresie genică. În acest fel, informația genetică primită de la părinți primește atât formă, cât și conținut, care este întruchipat în moștenitorul lor unic, feminin sau masculin.
Există o serie de patologii cu un tip de moștenire non-clasic, inclusiv sindromul Angelman, în care genele primite de la părinți ca parte a cromozomilor perechi poartă o amprentă unică a părinților și se manifestă în moduri diferite.
Așadar, sindromul Angelman este un exemplu frapant de amprentare genomică, în care expresia genelor în corpul copilului depinde direct de părintele de la care au fost primite alelele (forme diferite ale aceleiași gene, primite de la tată și mamă, situate pe secțiuni identice ale cromozomilor perechi). Adică, doar anomaliile cromozomului matern duc la dezvoltarea sindromului, în timp ce mutațiile și tulburările structurale ale cromozomului patern provoacă patologii complet diferite.
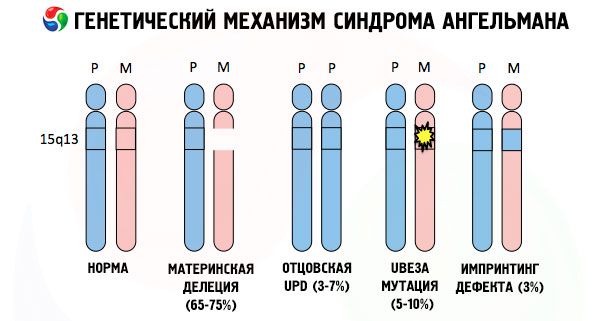
În această patologie, există o lipsă a anumitor gene în cromozomul matern sau o pierdere/reducere a activității unor gene individuale (în marea majoritate a cazurilor, gena ube3a, care este implicată în metabolismul ubiquitinei, o proteină ce reglează degradarea altor proteine). Drept urmare, copilul este diagnosticat cu anomalii de dezvoltare mentală și deformități fizice.
Simptome Sindromul Angelman
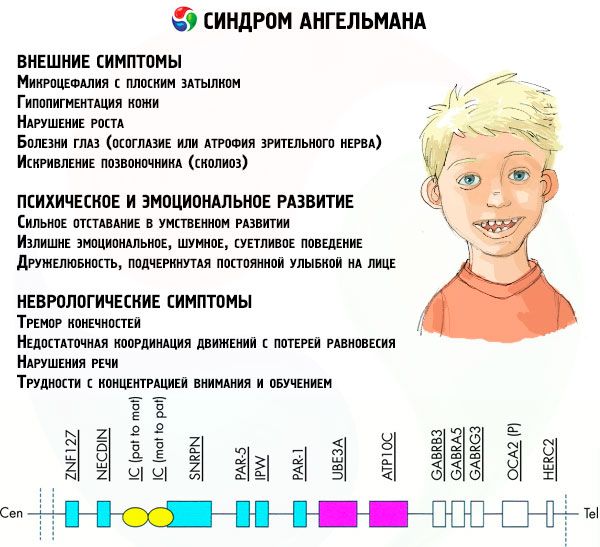
Simptomele sindromului Angelman afectează diverse aspecte ale vieții și dezvoltării unui copil: fizic, neurologic, mental. Pe baza acestui fapt, pot fi identificate 3 grupuri de simptome care indică dezvoltarea acestei patologii.
- Simptome externe sau fizice:
- un cap disproporționat de mic în comparație cu corpul și membrele, care au dimensiuni normale,
- gura prea largă,
- aproape întotdeauna există un zâmbet pe față (cu gura deschisă),
- dinți rari,
- buza superioară îngustă,
- limbă lată și proeminentă frecvent,
- maxilar inferior proeminent,
- bărbie ascuțită,
- piele foarte deschisă la culoare, adesea păr (albinism, asociat cu faptul că organismul nu produce pigmentul melanină),
- pete întunecate pe pielea deschisă la culoare (hipopigmentare din cauza producției insuficiente de melanină)
- simptome fizice sau externe: boli oculare precum strabismul sau atrofia nervului optic,
- curbura coloanei vertebrale (scolioză),
- picioare rigide (la mers, o persoană nu își îndoaie picioarele la genunchi din cauza mobilității reduse a articulațiilor, de unde și comparația cu mersul unei păpuși).
- Simptome legate de dezvoltarea mentală și emoțională:
- retard mintal sever,
- comportament excesiv de emoțional, zgomotos, agitat,
- bătăi frecvente din palme,
- a exprimat prietenie, subliniată de un zâmbet constant pe față,
- râs frecvent fără motiv.
- Simptome neurologice:
- tremorul membrelor,
- coordonare insuficientă a mișcărilor cu pierderea echilibrului,
- scăderea tonusului muscular,
- diverse tulburări de somn,
- crize isterice frecvente în copilărie,
- tulburări de vorbire (copilul începe să vorbească târziu, are abilități de comunicare slabe și vorbire neclară),
- hiperactivitate pe fondul unei excitabilități crescute,
- dificultăți de concentrare și învățare.
Dar aceasta este o imagine generalizată a bolii. De fapt, tabloul clinic al sindromului Angelman depinde în mare măsură de stadiul de dezvoltare al bolii și de tipul de mutație cromozomială care a cauzat patologia. Aceasta înseamnă că simptomele bolii pot diferi semnificativ la diferiți pacienți, ceea ce pentru o lungă perioadă de timp nu ne-a permis să distingem patologia de altele cu un tablou clinic similar.
Printre numărul total de simptome, putem evidenția pe cele care sunt caracteristice tuturor pacienților, fără excepție:
- retard mintal sever,
- comportament inadecvat (râs nejustificat, excitabilitate crescută, concentrare slabă, stare de euforie),
- subdezvoltarea abilităților motorii,
- coordonare deficitară a mișcărilor, ataxie la mers (ritm inegal, legănare dintr-o parte în alta etc.), tremor al membrelor.
- tulburare de dezvoltare a vorbirii cu predominanță a mijloacelor de comunicare nonverbale.
Printre simptomele întâlnite de marea majoritate a pacienților, se pot distinge următoarele:
- disproporția dintre cap și corp cauzată de dezvoltarea fizică întârziată,
- la mulți pacienți, forma craniului este de așa natură încât dimensiunea creierului rămâne mai mică decât la persoanele sănătoase (microcefalie),
- crize epileptice înainte de vârsta de 3 ani, cu o scădere progresivă a intensității și frecvenței la o vârstă mai înaintată,
- distorsiunea parametrilor EEG (fluctuații și amplitudine mare a undelor de joasă frecvență).
Aceste simptome sunt destul de frecvente, însă 20% dintre pacienții cu sindrom Angelman nu le prezintă.
Chiar mai rar, este posibil să se diagnosticheze astfel de manifestări ale bolii, cum ar fi:
- strabism sever sau ușor,
- control deficitar al mișcării limbii, ceea ce duce la faptul că pacienții scot adesea limba fără motiv,
- dificultăți la înghițire și supt, în special la copiii mici,
- perturbarea pigmentării pielii și a ochilor,
- brațele ridicate sau îndoite în timpul mersului,
- hiperreflexie,
- tulburări de somn, în special în copilărie,
- salivație frecventă,
- sete insațiabilă,
- mișcări de mestecare excesiv de active,
- hipersensibilitate la căldură,
- partea din spate plată a capului,
- maxilar inferior proeminent,
- palme netede.
Un procent destul de mare dintre pacienți au probleme cu urinarea, pe care le controlează slab, abilități motorii fine afectate, ceea ce creează dificultăți în îngrijirea personală și învățare, precum și exces de greutate. Aproape toți pacienții experimentează pubertatea mai târziu decât colegii sănătoși.
Copiii cu sindrom Angelman percep bine vorbirea orală și o înțeleg, dar nu vor să participe la conversații, limitându-și discursul la câteva zeci de cuvinte necesare în viața de zi cu zi. Cu toate acestea, la vârsta adultă, acești pacienți arată mai tineri decât colegii lor, fără patologii genetice.
Multe simptome ale sindromului Angelman sunt inconstante, așadar tabloul clinic al bolii se schimbă semnificativ odată cu vârsta. Convulsiile și crizele epileptice devin mai puțin frecvente sau dispar cu totul, pacientul devine mai puțin excitabil, iar somnul se îmbunătățește.
Complicații și consecințe
Sindromul Angelman este o patologie cromozomială severă, în prezent practic incurabilă, care îi privează pe pacienți de oportunitatea de a trăi o viață normală. Cum va fi viața unui copil cu AS depinde în mare măsură de tipul de anomalie cromozomială.
Duplicarea unui segment cromozomial este incompatibilă cu viața în majoritatea cazurilor. Și chiar dacă astfel de pacienți nu mor în copilărie și nu ajung la pubertate, nu au nicio șansă să aibă copii.
Ștergerea sau absența unei părți din genele care apar cel mai des în sindromul Angelman reprezintă un obstacol în calea învățării mersului și a vorbirii de către copil. Acești copii au o formă mai severă de retard mintal, iar crizele epileptice apar mai des, iar intensitatea lor este mult mai mare decât la pacienții cu alte anomalii cromozomiale.
Dacă există o mutație a unei singure gene, cu atenția și abordarea cuvenite, copilul poate fi învățat elementele de bază ale îngrijirii de sine, comunicării și interacțiunii într-un grup, deși va rămâne în urma colegilor săi în dezvoltare.
Pentru copiii cu sindrom Angelman, care sunt amabili din fire, cel mai important lucru este dragostea și atenția părinților lor. Numai în acest caz educația copilului va da roade, chiar dacă este mică. Desigur, pacienții cu AS nu vor putea studia într-o școală obișnuită. Au nevoie de clase speciale în care copiii vor fi învățați mai întâi să se concentreze, iar apoi, treptat, li se vor oferi elementele de bază ale cunoștințelor școlare.
Diagnostice Sindromul Angelman
Sindromul Angelman este o patologie congenitală a dezvoltării. Însă, din anumite circumstanțe, este adesea imposibil să fie diagnosticat în copilărie și în primii ani de viață. Acest lucru se datorează nespecificității și expresiei slabe a simptomelor la sugari și copii sub 3 ani. Iar prevalența bolii în țara noastră nu este atât de mare încât medicii să fi învățat să o recunoască printre semenii săi.
Sindromul Angelman la sugari se poate manifesta prin scăderea tonusului muscular, care se manifestă prin probleme de hrănire (slăbiciune a reflexului de supt și înghițire) și, ulterior, prin dificultăți în învățarea mersului (acești copii încep să meargă mult mai târziu). Aceste simptome sunt primele semne ale unei anomalii de dezvoltare la sugar, care poate fi asociată cu o anomalie cromozomială. Doar analiza genetică poate confirma această presupunere.
O atenție deosebită se acordă copiilor ai căror părinți au diverse afecțiuni genomice sau cromozomiale. La urma urmei, boala s-ar putea să nu se manifeste la început, iar dacă patologia este detectată la timp, începând să lucreze intens cu copilul, este posibil să se obțină un succes semnificativ mai mare în învățare, încetinind progresia bolii.
Dacă părinții prezintă diverse anomalii cromozomiale, analiza genetică se efectuează chiar înainte de nașterea bebelușului, deoarece SA este una dintre patologiile care pot fi detectate în stadiul embrionar.
Colectarea materialului pentru cercetarea genetică se poate realiza în două moduri:
- invaziv (cu un anumit procent de risc, deoarece este necesară penetrarea uterului pentru a preleva o mostră de lichid amniotic),
- neinvazivă (analiza ADN-ului bebelușului din sângele mamei).
Apoi se efectuează următoarele studii:
- hibridizare fluorescentă in situ (metoda FISH) – legarea unei sonde de ADN marcate cu un colorant special la ADN-ul studiat, urmată de examinare la microscop.
- analiza mutațiilor din gena ube3a și a genelor imprimate,
- Analiza metilației ADN-ului folosind metode speciale utilizate în genetică.
Testele genetice oferă informații destul de precise în cazul anomaliilor cromozomiale, ceea ce înseamnă că viitorii părinți știu dinainte la ce să se pregătească. Există însă și excepții. La un anumit grup de pacienți, în prezența tuturor simptomelor care indică patologia, rezultatele testelor rămân normale. Adică, patologia poate fi identificată doar prin observarea atentă a copilului încă din copilărie: cum mănâncă, când a început să meargă și să vorbească, dacă își îndoaie picioarele când merge etc.
Pe lângă metoda FISH, printre metodele instrumentale de diagnostic pentru sindromul Angelman, se poate evidenția tomografia (CT sau RMN), care ajută la determinarea stării și dimensiunii creierului, și electroencefalograma (EEG), care arată cum funcționează fiecare parte a creierului.
Medicii pun de obicei un diagnostic final la vârsta de 3-7 ani, când pacientul prezintă deja majoritatea simptomelor și dinamica dezvoltării bolii este vizibilă.
Ce teste sunt necesare?
Diagnostic diferentiat
Sindromul Angelman este o patologie genetică care practic nu are manifestări specifice. Majoritatea simptomelor pot indica în egală măsură atât AS, cât și alte patologii genetice.
Diagnosticul diferențial al sindromului Angelman se efectuează cu următoarele patologii:
- Sindromul Pitt-Hopkins (pacienții sunt caracterizați prin retard mintal, caracter vesel, zâmbitori, au o gură destul de mare și largă, se observă microcefalie). Diferența constă în atacurile de hiperventilație și ținerea respirației în stare de veghe.
- Sindromul Christianson (pacienții sunt persoane cu retard mintal, cu o dispoziție veselă, incapabile să vorbească, caracterizate prin microcefalie, ataxie, convulsii, mișcări musculare involuntare).
- Sindromul Mowat-Wilson (simptome: retard mintal, crize epileptice, bărbie ascuțită, gură deschisă, expresie veselă pe față, microcefalie). Distincție: distanță mare între ochi, ochi oblici spre interior, vârful rotunjit al nasului, auriculă întoarsă înapoi.
- Sindromul Kabuki (caracterizat prin retard mintal ușor până la moderat, probleme de vorbire și motorie, slăbiciune musculară, crize epileptice, microcefalie, intervale lungi între mâncărimi și coordonare deficitară). Caracterizat prin sprâncene arcuite, porțiunea laterală rătăcită a pleoapei inferioare, ochi depărtați, fisuri palpebrale lungi cu gene lungi și groase.
- Sindromul Rett (diferențiere de AS la femei). Simptome: întârziere în dezvoltarea vorbirii, convulsii, microcefalie. Diferența constă în lipsa unei expresii vesele pe față, apar atacuri de apnee și apraxie, care progresează în timp.
- Sindromul de retardare mintală autosomal recesiv 38 (simptome: retard mintal marcat cu întârzieri în abilitățile motorii și vorbire, slăbiciune musculară, probleme de hrănire în copilărie, impulsivitate). Trăsătura distinctivă este culoarea albastră a irisului.
- Sindromul de duplicație a genei MECP 2 (diferențiere de SA la bărbați). Simptome: retard mintal sever, slăbiciune musculară încă din copilărie, probleme de vorbire sau lipsa vorbirii, epilepsie. Distincții: miopatie progresivă, infecții recurente.
- Sindromul Kleefstra (simptome: probleme de vorbire și gândire, slăbiciune musculară, tulburări de somn, lipsă de atenție, gură deschisă, hiperactivitate, convulsii, ataxie, tulburări de echilibru). Trăsături distinctive: față plată, nas scurt și turtit, ochi mari și depărtați, buză inferioară mare și rătăcită, izbucniri agresive.
- Sindromul Smith-Magenis (caracterizat prin convulsii, probleme de somn, tulburări de dezvoltare intelectuală și motorie). Trăsăturile distinctive includ o față lată și plată și o frunte proeminentă.
- Sindromul Koolen-de Vries (retard mintal ușor până la moderat, slăbiciune musculară, convulsii, prietenie). Trăsături distinctive: față lungă cu frunte înaltă, urechi proeminente, ochi oblici, mobilitate articulară ridicată, malformații cardiace congenitale.
- Sindromul Phelan-McDermid (simptome: retard mintal, tulburări de vorbire sau lipsa vorbirii). Distincții: mâini mari cu mușchi dezvoltați, slăbiciune musculară de la naștere, transpirație slabă.
Patologii precum deficitul de adenilsuccinat, sindromul de retard mintal autosomal recesiv 1, sindromul de duplicație a cromozomului 2q23.1, sindroamele de haploinsuficiență genetică FOXG1, STXBP1 sau MEF2C și altele se pot „mândri” cu simptome similare sindromului Angelman.
Sarcina medicului este de a pune un diagnostic precis, diferențiind sindromul Angelman de patologii cu simptome similare și de a prescrie un tratament eficient, relevant pentru stadiul diagnosticat al bolii.
Cine să contactați?
Tratament Sindromul Angelman
Sindromul Angelman este una dintre acele patologii pentru care medicina încă caută un tratament eficient. Tratamentul etiologic al bolii se află în stadiul de dezvoltare prin diverse metode și mijloace, multe dintre ele nefiind încă testate pe oameni. Aceasta înseamnă că, deocamdată, medicii trebuie să se limiteze la terapia simptomatică, care ajută la ameliorarea situației dificile a copiilor și adulților cu sindromul marionetei, care suferă de crize epileptice, salivație, hipotensiune arterială și tulburări de somn.
Astfel, este posibilă reducerea frecvenței și intensității crizelor epileptice cu ajutorul unui medicament anticonvulsivant selectat corespunzător. Dar întreaga dificultate constă în faptul că crizele la pacienții cu SA diferă de crizele epileptice obișnuite prin faptul că sunt caracterizate de mai multe tipuri de crize, ceea ce înseamnă că afecțiunea poate fi ameliorată prin administrarea mai multor medicamente simultan.
Cele mai populare anticonvulsivante utilizate pentru tratamentul sindromului Angelman sunt: acidul valproic, topiramatul, lamotrigina, levetiracetamul, clonazepamul și medicamentele pe bază de acestea. Mai puțin utilizate sunt medicamentele pe bază de carmazepină, fenitoină, fenobarbital, etosuximidă, deoarece unele dintre ele pot provoca un efect paradoxal constând în intensificarea și creșterea frecvenței crizelor epileptice. Acest lucru se întâmplă dacă medicamentul este utilizat ca parte a monoterapiei.
Pentru tratarea salivației excesive, se utilizează de obicei două metode: medicamentoasă (medicamente care suprimă producția de salivă) și chirurgicală, care implică reimplantarea canalelor salivare. Însă, în cazul SA, aceste metode sunt considerate ineficiente, iar problema rămâne deschisă. Părinții și cei care îngrijesc astfel de pacienți trebuie să acorde o atenție deosebită acestei probleme, deoarece pacienții înșiși, de obicei, nu controlează salivația excesivă, iar unii pur și simplu nu sunt capabili să se îngrijească singuri.
O altă problemă este durata scurtă a somnului. Adesea, copiii cu sindrom Angelman nu dorm mai mult de 5 ore, ceea ce are un impact negativ asupra funcționării întregului corp. Copiii ușor excitabili, activi, cărora le plac jocurile și comunicarea (chiar dacă încearcă să se limiteze la metode nonverbale), sunt vizibil obosiți în timpul zilei. Pentru a se odihni bine, corpul are nevoie de un somn profund și complet, dar tocmai acesta este secretul.
Se pare că medicamentele sedative (fenotiazine și antipsihotice atipice) care calmează sistemul nervos ar trebui să fie suficiente pentru a îmbunătăți somnul la pacienții excitabili. Însă, în cazul sindromului angioneurotic de tip AS, utilizarea unor astfel de medicamente este plină de apariția unor efecte negative. Prin urmare, medicii preferă în continuare somniferele ușoare, cum ar fi Melatonina (un medicament hormonal natural bazat pe hormonul somnului), care se administrează pacienților cu o oră înainte de culcare, în cantitate de 1 comprimat, și Difenhidramina. Frecvența administrării și dozajul acestora sunt determinate de medic în funcție de starea și vârsta pacientului.
Uneori, pacienții cu sindrom Angelman au probleme cu digestia și scaunul. Puteți îmbunătăți scaunul cu laxative (de preferință pe bază de plante).
Sau puteți aborda problema diferit, așa cum au făcut medicii americani, bazându-vă pe unele metode de tratare a autismului, deoarece multe simptome caracteristice AS sunt caracteristice și autismului (impulsivitate, mișcări involuntare, acțiuni repetitive, deficit de atenție, probleme de comunicare etc.). S-a observat că introducerea hormonului secretină, care normalizează digestia și scaunul, are un efect pozitiv asupra atenției pacienților, iar oxitocina ajută la îmbunătățirea abilităților cognitive și a memoriei copilului și la corectarea comportamentului.
Este adevărat, hormonii singuri nu sunt suficienți, mai ales când vine vorba de copii. În sindromul Angelman, sunt indicate terapia comportamentală, lucrul cu un psiholog și un logoped (predând metode de comunicare non-verbală și limbajul semnelor). Educația acestor copii ar trebui să se bazeze pe un program individual cu participarea unor profesori special instruiți, a unui psiholog și a părinților. Din păcate, acest lucru nu este posibil peste tot, iar familiile rămân singure cu problema lor.
Întrucât mulți pacienți tineri cu AS suferă de tonus muscular scăzut și probleme articulare, se acordă multă atenție fizioterapiei. Cel mai adesea, medicii recurg la utilizarea aplicațiilor de parafină, electroforeză și terapie magnetică.
Masajul tonic activ și exercițiile speciale de antrenament fizic terapeutic îl vor ajuta pe copilul bolnav să se ridice pe picioare și să meargă cu încredere după un timp. Gimnastica acvatică este deosebit de utilă în acest sens, fiind recomandată pentru gimnastica acvatică în apă rece. Aceasta crește tonusul muscular și îl învață pe copil să-și controleze corpul și să-și coordoneze mișcările.
Tratament anticonvulsivant
Cel mai periculos simptom al sindromului Angelman sunt convulsiile similare cu cele ale epilepsiei. Acest simptom este observat la 80% dintre pacienți, ceea ce înseamnă că tuturor acestora trebuie să li se prescrie un tratament anticonvulsivant eficient.
Tratamentul crizelor epileptice se efectuează cu ajutorul vitaminelor și anticonvulsivantelor. În sindromul Angelman, însoțit de un sindrom convulsiv, vitaminele din grupa B, precum și vitaminele C, D și E vor fi utile. Dar prescrierea terapiei cu vitamine pe cont propriu în acest caz este foarte periculoasă, deoarece aportul necontrolat de vitamine poate reduce eficacitatea medicamentelor antiepileptice și poate provoca crize noi, mai severe și mai prelungite.
Selectarea medicamentelor anticonvulsivante și prescrierea dozei lor eficiente trebuie făcute, de asemenea, de către un medic specialist. Acesta decide, de asemenea, dacă un singur medicament va fi suficient sau dacă pacientul va trebui să ia 2 sau mai multe medicamente pentru o perioadă lungă de timp.
Pentru majoritatea pacienților, medicii prescriu medicamente cu acid valproic (acid valproic, Depakine, Convulex, Valparin etc.), care previn convulsiile și îmbunătățesc starea de spirit și starea mentală a pacienților.
Acidul valproic este disponibil sub formă de tablete, sirop și soluții injectabile. Cel mai popular medicament este medicamentul cu eliberare prelungită „Depakine” sub formă de tablete și ca soluție pentru administrare intravenoasă. Dozajul medicamentului este determinat individual de medic, în funcție de greutatea, vârsta și starea pacientului.
Medicamentul se administrează în timpul meselor de 2 până la 3 ori pe zi. Doza zilnică medie este de 20-30 mg per kilogram din greutatea pacientului, maximul fiind de 50 mg/kg pe zi.
Contraindicații pentru utilizare. Nu utilizați în caz de disfuncție hepatică și pancreatică, diateză hemoragică, hepatită, porfirie și hipersensibilitate la medicament.
Reacțiile adverse includ tremor al mâinilor, tulburări digestive și ale scaunului și modificări ale greutății corporale.
„Topiramatul” este, de asemenea, un medicament de elecție pentru AS. Este produs sub formă de tablete și este utilizat atât ca parte a monoterapiei, cât și în combinație cu alte medicamente.
Mod de administrare și dozaj. Comprimatele se administrează oral, indiferent de aportul alimentar. Doza zilnică inițială pentru adulți este de 25-50 mg, pentru copii - 0,5-1 mg/kg. În fiecare săptămână, doza se crește conform instrucțiunilor medicului.
Medicamentul nu trebuie administrat în timpul sarcinii și alăptării, precum și în caz de hipersensibilitate la componentele sale. Medicamentul are multe efecte secundare diferite.
Medicamente pe care un medic le poate prescrie pentru sindromul Angelman: Clomazepam, Rivotril, Lamotrigină, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra etc.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Medicina tradițională și homeopatia
Medicina tradițională, precum preparatele homeopate, este desigur relativ sigură, dar eficacitatea unui astfel de tratament pentru sindromul Angelman poate fi considerată controversată.
Deși tratamentul popular poate ajuta în continuare în anumite privințe. Vorbim despre oprirea crizelor epileptice. În acest sens, tratamentul pe bază de plante poate fi destul de eficient.
Un efect bun este asigurat de o colecție medicinală pe bază de bujor, lemn dulce și lintiță (componentele se iau în cantități egale). Plantele trebuie măcinate până se fac făină. După 2 săptămâni de la începerea administrării, puteți observa o scădere semnificativă a frecvenței convulsiilor.
Decoctul de lavandă (1 linguriță la un pahar de apă clocotită) este, de asemenea, util pentru crampe. Amestecul se fierbe timp de 5 minute și se lasă la infuzat o jumătate de oră. Medicamentul se ia seara, timp de 14 zile.
O infuzie apoasă (sau alcoolică) de gamba este considerată eficientă pentru crizele epileptice.
Dintre preparatele homeopate pentru prevenirea convulsiilor în sindromul Angelman, puteți utiliza medicamente pe bază de mușețel și motherwort, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Dar trebuie ținut cont de faptul că numai un medic homeopat poate prescrie doze eficiente și sigure de medicamente în fiecare caz specific.
Profilaxie
După cum probabil cititorul a înțeles deja, medicina nu este încă capabilă să prevină mutațiile genetice și alte anomalii cromozomiale, precum și să corecteze situația. Acest lucru se poate întâmpla oricui, deoarece copiii cu sindrom Angelman se nasc din părinți sănătoși, iar genetica, care este în prezent una dintre cele mai puțin studiate ramuri ale medicinei, nu poate încă explica acest lucru.
Singurul lucru care se poate face este să se adopte o abordare responsabilă a planificării sarcinii, să se înregistreze și să se supună la controale la timp. Dar, din nou, o astfel de măsură va fi mai degrabă educativă decât preventivă, ca orice controale. Însă părinții tineri vor ști dinainte la ce să se pregătească și, în cazul unui răspuns pozitiv, vor decide dacă își pot asuma o astfel de responsabilitate precum creșterea unui copil bolnav.
Prognoză
Prognosticul sindromului Angelman depinde de natura anomaliei cromozomiale și de promptitudinea detectării acesteia. Cei mai afectați sunt acei copii al căror cromozom 15 conține „lacune” în gene (deleții). Probabilitatea ca acești pacienți să meargă și să vorbească este extrem de scăzută. Alte cazuri pot fi corectate printr-o abordare atentă și cu dragoste pentru copilul dumneavoastră.
Din păcate, astfel de pacienți nu vor putea deveni membri cu drepturi depline ai societății, în ciuda faptului că sunt departe de a fi proști, ei înțeleg vorbirea și semnificația acesteia. Cu toate acestea, vor avea probleme de comunicare pentru tot restul vieții. Pacienților li se poate preda limbajul semnelor încă din copilărie, dar nu li se poate impune să comunice folosind cuvinte. Vocabularul pacienților „vorbitori” este limitat la minimul de cuvinte folosite în viața de zi cu zi (5-15 cuvinte).
În ceea ce privește speranța de viață și starea generală de sănătate a pacienților cu sindrom Angelman, cifrele fluctuează în jurul valorilor medii. La vârsta adultă, pacienții se confruntă în mare parte cu probleme de sănătate precum scolioza și obezitatea, care, cu abordarea corectă a tratamentului, nu pun viața în pericol.