Expert medical al articolului

Noile publicații
Alcaptonurie - patologie enzimatică congenitală
Ultima examinare: 28.11.2021

Tot conținutul iLive este revizuit din punct de vedere medical sau verificat pentru a vă asigura cât mai multă precizie de fapt.
Avem linii directoare de aprovizionare stricte și legătura numai cu site-uri cu reputație media, instituții de cercetare academică și, ori de câte ori este posibil, studii medicale revizuite de experți. Rețineți că numerele din paranteze ([1], [2], etc.) sunt link-uri clickabile la aceste studii.
Dacă considerați că oricare dintre conținuturile noastre este inexactă, depășită sau îndoielnică, selectați-o și apăsați pe Ctrl + Enter.
Una dintre foarte rare tulburări metabolice - alcaptonurie - se referă la anomalii congenitale în metabolismul aminoacizilor tirozină.
De asemenea, acest sindrom poate fi numit deficit de omogentizat oxidază, omogentizinurie, ocronoză ereditară sau boală de urină neagră. [1]
Epidemiologie
Conform statisticilor, nu există mai mult de nouă cazuri de alcaptonurie la 1 milion din populație. Și în majoritatea țărilor europene - un caz la 100-250 de mii de nașteri vii.
Dintre țările europene, excepția este Slovacia (în special regiunea relativ mică din nord-vest), unde prevalența alcaptonuriei este un caz la 19 mii de nou-născuți. După toate probabilitățile, acest lucru se datorează faptului că printre familiile romilor slovaci care locuiesc acolo, nivelul consangvinizării (căsătoriile între veri) este cel mai ridicat din Europa: 10-14%. [2]
Cauze alcaptonurie
Cauzele exacte ale alcaptonuriei, ca tulburare congenitală a catabolismului (descompunerea metabolică) a tirozinei α-aminoacidice aromatice (homociclice), au fost stabilite: acest tip de tulburare metabolică este o consecință a mutațiilor heterozigote homozigote sau complexe de una din mii de gene de pe cromozomul 3, mai exact, gena HqG21 la locus 3 -q23 pe brațul lung al cromozomului. Această genă codifică secvențele nucleotidice ale enzimei hepatice homogentizat-1,2-dioxigenază [3](numită și oxid acid acid homogentizic sau homogentizat oxidază), o metaloproteină care conține fier necesară pentru una dintre etapele de descompunere a tirozinei în organism. [4], [5]
Astfel, alcaptonuria este un defect în enzima homogentizat-1,2-dioxigenază, mai precis, rezultatul deficienței sale determinate genetic sau a absenței complete. [6]
Fiind o fermentopatie congenitală, alcaptonuria este moștenită ca o trăsătură autosomală recesivă, adică pentru ca alcaptonuria să apară la copii, ambii părinți trebuie să aibă o genă modificată pentru această enzimă, deoarece fiecare dintre ei transmite copilului o singură copie a genă din cele două disponibile.
Conform celor mai recente date, există mai mult de două sute de variante ale modificării genei HGD și se observă cel mai adesea mutații, translocație și splicing.
Factori de risc
Singurul factor de risc pentru dezvoltarea acestei fermentopatii congenitale este istoricul familial și moștenirea a două copii modificate ale genei HGD, dacă părinții nu prezintă alcaptonurie (riscul de transmitere a anomaliei este de 25%) sau unul dintre părinții au această tulburare. [7]
Patogeneza
Cel mai important rol joacă tirosina în sinteza proteinelor, producerea de cromoproteine - melanina pigmentară a pielii, precum și a hormonilor tiroidieni și a neurotransmițătorilor de catecolamină.
Mecanismul de reglare a cantității de tirozină din celule este foarte complex, iar corpul își normalizează conținutul în exces prin divizare. Procesul de catabolism al tirozinei, ca toți aminoacizii aromatici, este multistep și se desfășoară în mai multe etape. Mai mult, fiecare etapă a descompunerii metabolice a tirozinei are loc cu participarea unei anumite enzime și formarea unui compus intermediar.
Deci, mai întâi, aminoacidul este scindat de para-hidroxifenilpiruvat, care se transformă într-o alcaponă - acid 2,5-dihidroxifenilacetic sau acid homogentizic. Mai mult, alcapona ar trebui transformată în acid maleacetic, dar acest lucru nu se întâmplă. [8]
Și patogeneza alcaptonuriei constă în oprirea reacțiilor biochimice ale catabolismului tirozinei în stadiul de formare a acidului homogentizic: pur și simplu nu există o enzimă necesară pentru a-l descompune - homogentizați oxidaza.
Acidul homogentizic nu este utilizat de organism și se poate acumula cu excreție prin rinichi. În plus, este oxidat la benzoquinoacetat (acid benzoquinoneacetic), care, prin legarea cu moleculele de țesuturi și fluide corporale, formează compuși biopolimeri colorați ca melanina.
Acumularea acestor produse intermediare în țesut duce la întreruperea structurii de colagen a țesutului cartilaginos, datorită căreia elasticitatea acestuia scade - odată cu apariția multor semne clinice de alcaptonurie și dezvoltarea complicațiilor. [9]
Simptome alcaptonurie
Alcaptonuria la nou-născuți și sugari se manifestă prin întunecarea urinei. La contactul cu aerul, culoarea urinei de pe scutece, scutece și lenjerie de corp devine maro închis; acest lucru se datorează acumulării și eliberării de acid homogentizic, care este oxidat în benzoquinoacetat. [10]
În absența altor simptome, alcaptonuria la copii la o vârstă fragedă nu este adesea recunoscută în timp util, deoarece după urinare urina se poate întuneca după câteva ore. Potrivit unor rapoarte, în condițiile clinicilor, sunt detectate doar o cincime din copiii cu vârsta sub 12 luni care s-au născut cu această fermentopatie. Prin urmare, este foarte important ca părinții să fie atenți la îngrijirea bebelușului.
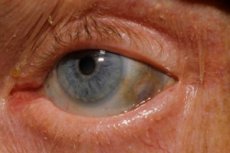
În plus, semnele timpurii includ pigmentarea (gri albăstrui) a sclerei ochiului și a cartilajului auriculelor și nasului, adesea denumită ocronoză. [11]
În timp, apar alte simptome:
- pigmentare severă a pielii pe pomeți, axile și organele genitale;
- vopsirea hainelor la contactul cu părți ale corpului transpirate;
- atacuri de slăbiciune generală;
- voce ragusita.
Trebuie avut în vedere faptul că alcaptonuria și ochroza , așa cum sa menționat mai sus, sunt nume sinonime pentru aceeași încălcare a catabolismului tirozinei.
Boala siropului de arțar și alcaptonurie. Boala congenitală a siropului de arțar sau leucinoza se referă, de asemenea, la tulburări metabolice, are același tip de moștenire și chiar mutații apar pe același cromozom, dar se referă la gena care codifică complexul enzimatic al a-cetoacizilor ramificați dehidrogenază. Din această cauză, organismul nu poate descompune anumite componente proteice, în special aminoacizii leucina, izoleucina și valina. În această stare, urina (și ceara de urechi) are un miros dulce; în plus, în tabloul clinic al acestui tip de acidemie organică, se observă hipopigmentare, fluctuații ale tensiunii arteriale, convulsii, vărsături și diaree, o scădere a glicemiei, cetoacidoză, halucinații etc. Rata mortalității la copii este destul de mare. înalt; la adulții fără tratament, coma și decesul pot apărea din cauza edemului cerebral.
Albinismul și alcaptonuria „unesc” doar tirozina. Albinismul , inclusiv albinismul oculocutanat, este cauzat de mutații genetice care afectează producția pigmentului de melanină. Modificările congenitale sunt observate în gena TYR de pe cromozomul 11 (11q14.3), care codifică tirozinaza, o enzimă care conține cupru în melanozomi care este necesară pentru formarea pigmentului pielii pe baza produselor metabolismului tirozinei. Această boală este mult mai frecventă decât alcaptonuria.
Complicații și consecințe
Consecințele și complicațiile alcaptonuriei cauzate de acțiunea metaboliților intermediari ai tirozinei - acizi homogentizici și benzoquinoneacetici - apar datorită depunerii de polimeri pigmentați reactivi, distrugerii fibrilelor de colagen și deteriorării stării cartilajului (cu o scădere a rezistenței acestora la solicitări mecanice).
Ani mai târziu, la vârsta adultă, se dezvoltă artrita degenerativă și osteoartrita articulațiilor mari (șold, sacroiliac și genunchi); îngustarea spațiilor intervertebrale (în special a coloanei lombare și toracice) - cu calcificare și formarea de osteofite; densitatea țesutului plăcilor osoase subcondrale scade, iar oasele subiacente pot suferi o remodelare patologică cu formarea de creșteri și deformare. [12]
Pot apărea leziuni ale valvelor cardiace (aortice și mitrale) și ale arterelor coronare, cu semne de boli coronariene, precum și formarea de pietre la rinichi și prostată, datorită aceleiași calcificări. [13], [14]
Diagnostice alcaptonurie
De obicei, diagnosticul tulburărilor metabolice congenitale se bazează pe studiul fluidelor corporale.
Pe baza a ce teste și ce reacții poate fi diagnosticată alcaptonurie? Sunt necesare teste de urină - pentru detectarea acidului homogentisic și determinarea nivelului acestuia (normal - 20-30 mg pe zi, crescut - 3-8 g). O probă de urină este examinată prin cromatografie de gaze sau spectrometrie de masă utilizând cromatografie lichidă; este posibil un test de screening pentru prezența clorurii ferice în urină. [15]
Există, de asemenea, o metodă rapidă de diagnostic - determinarea alcaptonului în pete uscate de urină pe hârtie (după intensitatea culorii).
La clarificarea diagnosticului, diagnosticul instrumental (raze X) implică identificarea semnelor cu raze X ale osteoartritei și a altor patologii articulare la pacienți.
Diagnosticul este confirmat de& metode genetice moleculare precum diagnosticul bolilor ereditare , cum ar fi testarea genetică și secvențierea ADN-ului. [16]
Diagnostic diferentiat
Diagnosticul diferențial include hemocromatoză și insuficiență hepatică acută a nou-născuților, melaninurie, porfirie acută intermitentă, limfohistiocitoză hemofagocitară, patologie mitocondrială primară, artrită reumatoidă, spondilită anchilozantă.
Cine să contactați?
Tratament alcaptonurie
Tratamentul principal pentru alcaptonurie este ingestia de doze mari (cel puțin 1000 mg pe zi) de acid ascorbic. La copii, acest lucru mărește excreția acidului homogentizic în urină, iar la adulți reduce nivelul derivatului său, acid benzoquinoneacetic, în urină și încetinește legarea acestuia de structurile țesutului conjunctiv al articulațiilor și colagenului. [17]
În clinicile din Europa de Vest, se testează medicamentul Nitizinonă (Orfalin), un medicament al unui grup de metaboliți care inhibă a doua etapă a catabolismului tirozinei: transformarea para-hidroxifenilpiruvatului în acid homogentizic. Cu toate acestea, utilizarea acestui agent farmacologic duce la acumularea de tirozină și poate provoca reacții adverse grave, inclusiv opacitatea și fotofobia corneei, sângerări la nivelul nasului și stomacului, insuficiență hepatică, modificări ale sângelui etc. Cu toate acestea, în Statele Unite, Nithizinone este aprobat de FDA pentru tratamentul tirozinemiei de tip I. [18], [19]
Ca atare, fizioterapia - terapia exercițiilor fizice pentru creșterea forței musculare și creșterea mobilității articulațiilor, balneo și peloidoterapia pentru limitarea durerii - se efectuează pentru problemele articulare cauzate de alcaptonurie.
Deși tirozina nu este furnizată numai cu alimente, ci și produsă în organism, pacienților cu alcaptonurie li se recomandă o dietă săracă în proteine și să limiteze aportul de alimente bogate în tirozină, în principal carne de vită și porc, produse lactate (în special brânzeturi), leguminoase, nuci etc. Semințe.
Profilaxie
Prevenirea mutațiilor genetice este imposibilă și pentru a preveni nașterea copiilor cu risc crescut de tulburări congenitale, există consiliere medicală și genetică, care este necesară înainte de sarcina planificată a acelor cupluri căsătorite în ale căror antecedente familiale există boli ale unui ereditar. Natură. [20]
Prognoză
Există foarte puține decese în cursul alcaptonuriei, iar moartea poate fi cauzată de complicații grave care afectează inima și rinichii. Deci, pentru speranța totală de viață a persoanelor cu alcaptonurie, prognosticul este favorabil.
Dar calitatea vieții este redusă datorită durerii intense la nivelul articulațiilor sau coloanei vertebrale cu limitare semnificativă a mobilității, adesea progresivă.